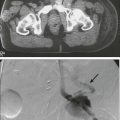
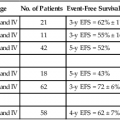
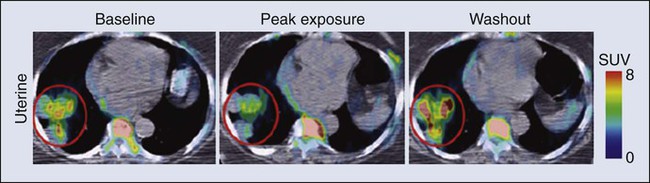
• Cancer pharmacology encompasses the spectrum of therapeutic issues from everyday clinical treatment to the earliest stages of new drug discovery. • The number of drugs approved for treating patients with cancer is rapidly rising. • The era of oral drugs for cancer treatment is well underway. • Drug actions, both desired anticancer effects and unfortunate adverse effects, are informed by the discipline of pharmacodynamics, which includes efforts to customize treatment for the specific tumor pathways in patients with cancer. • Drug delivery issues, including selection and adjustment of doses, routes, and schedules of administration, are addressed by the discipline of pharmacokinetics. • Navigating the explosion of information about cancer biology and the specific properties of drugs for cancer treatment will continue to be a challenge for oncologists and related health professionals. One of the most obvious metrics for the importance of cancer pharmacology is the rapid and continuing expansion in the number of drugs available to treat cancer. More than 100 agents are marketed for oncology, including 11 that were approved by the Food and Drug Administration (FDA) in 2012 (Box 29-1). As a point of reference, at the first meeting of the American Society for Clinical Oncology in 1964, only 12 drugs were marketed for oncology. Although this chapter focuses on small, synthetic molecules, one of the most exciting developments in cancer pharmacology is the successful transition of antibodies from the research stage to full integration with small molecules in the everyday treatment of patients. An increasing role is also developing for linkage of toxins that are guided to tumors by antibodies (see Chapter 32, “Therapeutic Antibodies and Immunologic Conjugates”). Although very different in terms of their size and manufacturing techniques, the principles of development for these new therapeutic classes have the same goals as for small molecules—for example, modulation of specific molecular targets and linkage to a diagnostic test for that target. Trastuzumab embodies all of these elements, including its extension into the realm of antibody-drug conjugates with the 2013 FDA approval of ado-trastuzumab emtansine. Genetics is a fundamental part of the study of cancer research (see Chapter 1, “Molecular Tools in Cancer Research”). The range of applications for genetics in cancer include hereditary predisposition and interactions of heredity with nonhereditary factors (see Chapter 13, “Genetic and Epigenetic Changes in Cancer”). As described in the preceding chapter (“Therapeutic Targeting of Cancer Cells”) and elsewhere (e.g., Chapter 8, “Vascular and Interstitial Biology of Tumors”), our understanding of the physiology and molecular biology of tumors has provided a wealth of potential targets for anticancer therapy. Although the relative intensity has magnified, the concept of molecular targeting and linkage to diagnostics for selection and monitoring of individual patients has a long history. The selection of hormonal therapies for patients whose tumors express the estrogen receptor was among the first successful uses of molecular targeting. Among several recent successful examples in the current era of targeted cancer pharmacology, two noteworthy cases are the approval and everyday use of imatinib only in patients with chronic myelogenous leukemia (CML) that is Philadelphia chromosome–positive and trastuzumab only in patients with HER2-positive tumors. In addition to the obvious attraction of matching drugs to the molecular characteristics of the tumor, the therapeutic index can also be improved by examining host tissues. In his last major research publication,1 Dr. Abeloff was a leader in a large multicenter study of the PG for cyclophosphamide. The goal was to determine if the germline DNA of patients altered the toxicity profile of cyclophosphamide. A subgroup of women was found who had variant GSTP1 alleles that were associated with less susceptibility to adverse hematologic toxicity in regimens containing cyclophosphamide. Another large cooperative group clinical study examined germline determinants of paclitaxel neurotoxicity.2 Wheeler et al.2 used genome-wide analyses coupled with well-characterized data from a cell bank to support a polygenic approach to understanding the variation in host tissue sensitivity to the sensory peripheral neuropathy associated with paclitaxel. As described in the various chapters relating to specific malignancies in Part III of this book, it is rare for a drug to be used as a single agent. The development of combinations would be an extensive topic in itself. Box 29-2 provides a set of points to consider for combinations. Within a combination chemotherapy regimen, the drugs are intended to interact at the PD level. Originally, a major strategy was to combine cancer drugs with nonoverlapping toxicities. Increasingly, more detailed knowledge regarding pathways for cancer drugs permits the design and evaluation of combination strategies that target parallel and/or sequential pathways. Most combinations are based on a preexisting scientific rationale or by addition of a new agent to an established regimen. However, it would be presumptuous to assume that all possibilities are already understood. One tool to expand the scope of hypothesis generation is the systematic study of all cancer drugs in combination with each other, or in combination with approved agents outside oncology, or testing every investigational agent versus all approved cancer drugs.3 Although this screening approach begins as an empiric exercise, the successful combinations become challenges for explaining the underlying mechanisms of action. The focus on drugs that are already approved provides a potentially fast route to clinical implementation. With the widening availability of fluorine-18 fluorodeoxyglucose (18F-FDG), a consensus was building in the late 1990s that positron emission tomography (PET) would emerge as a complementary tool to anatomic imaging modalities.4 Since then, FDG has been highly successful as a general probe in many tumor types, providing additional information to help separate malignant from benign lesions and to monitor response to therapy. FDG is now an approved drug for imaging. Its success has spurred interest in the development of other probes for PET imaging that are currently in the investigational stage of clinical evaluation, including 18F-fluorothymidine (FLT). Figure 29-1 shows the results for FLT in a patient with uterine cancer.5 At baseline, the FLT image shows major uptake by the tumor, indicative of active proliferation. During treatment with sunitinib, uptake of FLT in the tumor decreased, which was interpreted as reduced proliferation. The image following the withdrawal of treatment exhibited a flare, that is, intensity that rebounded above the baseline value. Anatomic imaging by computed tomography (CT) readily identified the tumor and could monitor regression if that occurred, but FLT provided the ability to follow molecular pathways in real time. Clearance is at the top of the list for drug delivery parameters in Box 29-3 because it is the factor with the greatest impact on dose-adjustment in cancer drug delivery. As stated in Box 29-4, clearance is the summation of all mechanisms for a drug to be removed from the systemic circulation, sometimes called total body clearance. Several specific applications of clearance for dosage adjustment will be covered in the next few sections. Compared with other medical areas in which fixed doses are prevalent, dosing in cancer pharmacology has long been titrated on the basis of each patient’s body weight or body surface area. These measurements are intended to serve as first approximations for clearance in the absence of data, but their incremental value compared with a standard dose is unfortunately small, and most current trials have adopted fixed doses, that is, “flat dosing.”6 The role of clearance in dosage adjustment is summarized in Box 29-4. The premise is that the standard dose was selected on the basis of the needs of patients with average drug clearance. If a patient has lower clearance than the average, then using the standard dose will lead to higher than average systemic exposure, with the probability of higher than average toxicity. Using a dose reduction to match the lower clearance will normalize the exposure of this patient relative to the average in the population. Similarly, a patient with higher than average clearance will have lower systemic drug exposure than will patients with the average exposure for the population. Although this lower systemic drug exposure will probably reduce the frequency or severity of toxicity, it has the potentially lethal consequence of reducing the response of the tumor. This overall strategy for dosage adjustment has been elegantly demonstrated for the first scenario in Box 29-3, namely, patients with impaired renal function. The seminal clinical study of carboplatin by Egorin et al.7 in patients with variable renal function remains the prototype for dosage adjustment based on impaired organ function. For the other two areas in Box 29-3 (drug-drug interactions and pharmacogenetic factors), many practical cases of dosage adjustment exist. Specific examples will be discussed in the following sections. Drug-drug metabolic interactions are possible for parenteral routes of drug delivery, but they are far more common for the oral route. Because metabolism is the primary determinant of clearance for most drugs, it is the dominant controller for changes in plasma concentrations. Following the paradigm in Box 29-4, inhibition of drug metabolism via a drug-drug interaction will produce lower clearance and thus higher systemic exposure. A reduction in dose may be necessary to avoid increased toxicity. Conversely, drugs such as phenytoin can induce the expression of certain metabolizing enzymes, which would decrease systemic concentrations for the drugs cleared via those enzymes, because the rate of their metabolism would be faster. Decreased exposure increases the probability of inadequate delivery of the cancer drug to the tumor. In the section on Oral Cancer Drugs, Table 29-1 provides some examples of the enzymatic pathways that metabolize cancer drugs. Table 29-1 Interaction of Drugs with Enzymes and Transporters for Recently Approved Oral Cancer Drugs *The drug metabolism pathways are: 1A2 = CYP1A2; 3A = CYP3A; 2D6 = CYP2D6; and various members of the CYP2C family: 2C8, 2C9, 2C19. For transport, the ABCB1 (MDR1) pathway is cited. Data retrieved from Food and Drug Administration approved labeling, found at http://www.accessdata.fda.gov/scripts/cder/drugsatfda/. In cancer pharmacology, the primary focus for transport of drugs has been the flux across the cell membranes of tumors. A major mechanism of drug resistance has been described for tumors with high expression of efflux pumps that reduce the availability of cancer drugs to their targets. Investigations into the influx of cancer drugs into tumors have also been conducted. White et al.8 demonstrated that patients with CML had more long-term benefit from imatinib if expression of its influx pump, OCT-1, was higher. In addition to the interactions of cancer drugs with transporters in the tumor, interactions occur among cancer drugs and cellular transport systems in host tissues that control uptake and elimination of drugs from the GI tract, the liver, and the kidneys. The expression of transporter function can be modulated by other drugs in a manner similar to drug-drug metabolic interactions. Indeed, some of the same inducers of metabolism (phenytoin or rifampin) are also major inducers of transporters. In contrast, ketoconazole, itraconazole, and certain psychopharmacologic agents are inhibitors. This focus on drug transport is important regardless of the route of drug delivery, but similar to metabolic interactions, additional factors exist that are related to control of absorption of cancer drugs from the GI tract. Transporters are also the underpinning of the blood-brain barrier and control the entry of cancer drugs into the brain to treat metastases or primary brain tumors.9 The source of drugs for patients with cancer is not restricted to those prescribed by oncologists, or even medicines prescribed by other medical practitioners. Self-medication with over-the-counter drugs and alternative therapies is prevalent in patients with cancer and includes substances that interact with cancer drugs (see Chapter 33, “Complementary and Alternative Medicine”). Parenteral administration will continue to be important, both for the many legacy drugs that remain part of regiments with established therapeutic value and for occasional cases in which an agent cannot be successfully delivered via the oral route. However, based on the up-front goal to achieve oral delivery for most new cancer drugs under development, especially for chronic daily dosing, it is easy to project that the ratio of oral to IV cancer drugs will continue to rise. As shown in Box 29-1, the FDA approved 11 cancer drugs in 2012. Overall, 7 of these 11 drugs are administered orally, including 7 of the 9 small synthetic compounds. As indicated in Box 29-5, adherence to the prescribed regimen is one of the first questions to explore. Although patients with cancer are highly motivated to carefully follow their dosing instructions, it is naïve to expect complete compliance and especially to ignore the problems of persistence over a long period. Marin et al.10 concluded that poor adherence may be the predominant reason for failure to achieve adequate molecular responses with imatinib. Partridge et al.11 examined the complex scenario of elderly patients with cancer, a population that has a high frequency of polypharmacy and perhaps a fading memory. For adjuvant therapy of early stage breast cancer with tamoxifen, 25% of patients took fewer than 80% of the expected doses, which is problematic. This study included no patients younger than 65 years, but the authors reported that adherence was not dependent on age for persons ranging from 65 to 89 years. Systemic drug exposure ratio, at equal doses: Although ease of oral cancer drug administration is generally a major advantage, it can become a liability when co-morbid conditions make swallowing drugs a major problem (see Chapter 43, “Oral Complications.” Cancer chemotherapy can produce emesis (see Chapter 42, “Nausea and Vomiting”), which thwarts the reliability of retaining swallowed doses. In addition, the treatment of these complications of cancer therapy can include the use of additional drugs to complicate the drug-drug interaction scenarios. For the representative set of 10 cancer drugs described in Table 29-2, the advice for four drugs (ruxolitinib, vemurafenib, crizotinib, and enzalutamide) is that the timing of food intake is not important. The other six drugs are split evenly between recommendations to take while fasting versus taking with a meal. For two of these six drugs, the magnitude of the food effect is provided. Table 29-2 Impact of Food on Drug Absorption for Recently Approved Oral Oncology Drugs* *If no preference is provided for taking with food or fasting, ± is indicated, and the effect size is not significant (NS). If no advice is provided, ?? is indicated. Data retrieved from Food and Drug Administration approved labeling, found at http://www.accessdata.fda.gov/scripts/cder/drugsatfda/. If these dosing recommendations are unintentionally or intentionally reversed, potentially serious issues of reduced efficacy or increased toxicity occur, along with potential economic issues. The fourfold increase in absorption of lapatinib with food has been the source of substantial public discussion,12 which has been extended to cover other oral cancer drugs.13 For abiraterone, the tenfold difference in absorption is sufficiently large to capture everyone’s attention. Perhaps abiraterone has a wider therapeutic index than most cancer drugs, but a tenfold difference in exposure seems unlikely to be without consequences. In addition to the general effects of food on solubility of drugs and passage into the body, a few food substances have highly specific effects on cancer drug metabolism and cancer drug transporters. The most notable example is the interaction of grapefruit juice with metabolism via the CYP3A family and the ABC family of transporters.14 The specificity of effects with grapefruit and drugs closely resembles the drug-drug interactions described later. For the same set of 10 representative oral cancer drugs described in Table 29-2 for food effects, Table 29-1 provides information from the FDA Web site for drug interactions with enzymes and transporters. The entries in Table 29-1 indicate that all of these drugs have at least some information about enzymatic pathways. The largest category of potential interactions is with the CYP3A family of enzymes, which are present in both intestines and liver and have potential interactions with 9 of the 10 drugs. Scattered information is presented for other members of the CYP superfamily: 1A2, 2C8, 2C9, 2C19, and 2D6. Interest is expanding rapidly in the role of transporters15 interacting with cancer drugs, but only one drug in this set, lapatinib, has information about the transporter pathways. Intraarterial catheterization has been technically feasible for delivery of cancer drugs to a variety of places in the body. The use of an implanted pump for the intraarterial delivery of fluorodeoxyuridine is approved for the treatment of liver metastases from colorectal cancer and is the most common application of this approach.16 Intraperitoneal delivery of drugs for treatment of localized ovarian cancer and other intraperitoneal malignancies has undergone many evaluations. Three randomized phase 3 studies have shown a survival benefit for intraperitoneal delivery compared with the IV route.17 Despite this success, the lack of familiarity with the catheters, uniform distribution within the peritoneal cavity, and other factors have slowed the penetration of this modality into routine practice. Speculation that many shortages could be bypassed by substituting drugs that are “almost as good” was dramatically dashed by the study of Metzger et al.18 Because of a lack of supply of mechlorethamine, cyclophosphamide was substituted in the multidrug regimen for children with Hodgkin lymphoma. In only 2 years, more than a doubling occurred in the number of patients who no longer had event-free survival. Within the context of recent trends for new cancer drugs (Box 29-6), the processes for development and discovery need to be clearly understood. However, the actual research that comprises these phases needs to adapt to the continuous stream of new findings in cancer biology. Further, the optimal approach to information gathering during human evaluation of cancer drugs is continually reevaluated, and the relevance of preclinical models to the clinical realities is continually improved. As indicated in Box 29-7, the clinical stages of drug development have been divided into phases that are related to the primary goals. These stages have historical and regulatory significance, but the boundaries are always being pushed, and it is not possible to assign every compound into the same bins. For patients with cancer, this view of clinical development is continually challenged by the compelling need to provide therapeutic options when no treatments have been established. The boundaries that are calibrated for less serious diseases may be a bit more flexible in oncology, just as the definition of maximum tolerated dose is a relative term that depends on the medical scenario. Phase 1, phase 2, and phase 3 are long-established terms. Phase 4 was created by a formal change in FDA regulations. Phase 0 is a less-formal term used by some investigators to describe early explorations to seek information before making a decision to launch full-scale development. Phase 2 clinical trials are the setting for determining if an agent or combination of agents has sufficient activity in well-defined groups of patients who have no established treatments.19 In the majority of phase 2 studies, there is only a single treatment group, but randomized designs of two or more groups have been increasingly used. The goal is to determine if the activity of the agent or combination is sufficient to move into much larger phase 3 trials with comparison to standard treatments. Unfortunately, areas within oncology remain for which no standard treatment has been established. In such cases, phase 2 studies can be sufficient for regulatory approval. Phase 0 clinical studies are an approach to first-in-human studies that extends the preclinical search for specific types of information into the clinic. Frequently, there are concentrations that are considered essential to activity.20,21 For oral therapy, concerns exist about the consistency of absorption. With the large and increasing number of drugs available for use (see the Appendix)22, it is not feasible to remember all the details about each one. The widespread availability and use of online resources, including FDA databases, supplements traditional textbooks. Assembling the key pieces of data is only the first step. Professional interpretation and judgment remain as central features.
Cancer Pharmacology
Introduction
Fundamental Science
Principles of Cancer Drug Action
Combinations of Drugs
Molecular Imaging of Cancer Drug Action
Principles of Cancer Drug Delivery
Dosage Adjustment Based on Clearance
Drug-Drug Interactions
Drug
Is This Drug Affected by Inducers/Inhibitors?
Does This Drug Cause Change for Other Drugs?*
Imatinib
3A
3A, 2D6
Lapatinib
3A
3A, 2D6, ABCB1
Pazopanib
3A
3A, 2D6, 2C8
Ruxolitinib
3A
Not reported
Vemurafenib
No data
3A, 1A2, 2D6
Crizotinib
3A
3A
Abiraterone
No data
2D6
Bosutinib
3A
No data
Regorafenib
3A
No data
Enzalutamide
2C8, 3A
3A, 2C9, 2C19
Drug-Drug Transporter Interactions
Interactions Via Self-Medication
Oral Cancer Drugs
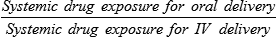
Drug
Whether Drugs Should Be Taken with Food
Size of Effect
Imatinib
Yes
??
Lapatinib
No
Fourfold
Pazopanib
No
??
Ruxolitinib
±
NS
Vemurafenib
±
NS
Crizotinib
±
NS
Abiraterone
No
Tenfold
Bosutinib
Yes
??
Regorafenib
Yes
??
Enzalutamide
±
NS
Clinical Relevance and Applications
Regional Cancer Drug Delivery
Shortages of Cancer Drugs
Development and Discovery of Cancer Drugs
Clinical Phases of Drug Development
What the Future Holds
Drug Name
Drug Class and/or Mechanism
Pharmacokinetics/Metabolism
Toxicity
Indications
Dosing
Abiraterone acetate (Zytiga)
Inhibits androgen biosynthesis
Do not take with food; inhibits CYP2D6
Joint swelling or discomfort; edema; monitor liver enzymes
Metastatic prostate cancer; castration resistant with prior use of docetaxel
1000 mg once a day orally with 5 mg prednisone BID
Ziv-aflibercept (Zaltrap)
Fusion protein binding VEGF and related ligands
No studies for renal, hepatic, or drug-drug interactions
Hemorrhage, GI perforation, wound healing problems, fistula, hypertension, arterial thrombosis, proteinuria
Metastatic colorectal cancer resistant or progressed while taking oxaliplatin
4 mg/kg via 1-hr IV (not bolus) every 2 wk
Altretamine (Hexalen), hexamethylmelamine, HMM
Alkylating agent
Well absorbed by mouth, metabolized in the liver; half-life 4-13 hr; metabolism may be slowed by cimetidine or enhanced by phenobarbital
Myelosuppression is dose limiting; leukopenia, thrombocytopenia, nausea, and vomiting are common; neurologic toxicity, including confusion, lethargy, weakness, and sensory changes, is common
Refractory ovarian carcinoma
4-12 mg/kg/day in divided doses for 3-6 wk or 150 mg/m2/day for 14 days each cycle; higher doses have been used
Amifostine (Ethyol), WR- 2721, ethiofos
Cytoprotectant; free-radical scavenger
After IV infusion, the drug is metabolized to a thiol metabolite, which is responsible for its beneficial activity
Transient hypotension is dose limiting; nausea, vomiting, and somnolence are common; sneezing, hypocalcemia, and flushing can be seen
Pretreatment with cisplatin; useful as a bone marrow, kidney, and nerve cytoprotectant; useful with other alkylators as well; also FDA approved as a radiation protectant to reduce xerostomia
740 mg/m2 IV infusion over 15 min given 15-30 min before the cytotoxic agent or radiation; lower doses and subcutaneous administration have also been used
Anastrazole (Arimidex)
Nonsteroidal aromatase inhibitor; blocks estrogen production selectively
Well absorbed from GI tract, maximum plasma levels within 2 hr; half-life is 50 hr; extensively metabolized in liver; despite hepatic and renal clearance being important, no adjustments needed for abnormal function of these organs because of the wide therapeutic index of this drug
Very well tolerated; asthenia, headache, and hot flashes occur in fewer than 15% of women; diarrhea, abdominal pain, anorexia, nausea, and vomiting occur in 10% or fewer; thrombophlebitis has been reported
Indications: As adjuvant therapy of breast cancer and for treatment of postmenopausal women with breast carcinoma who have progressed while being treated with tamoxifen
1 mg PO every day; higher doses are no more effective
Arsenic trioxide (Trisenox)
Novel arsenical differentiating agent
Half-life of this compound is unknown; it is methylated in the liver and eliminated in the urine
The “differentiation syndrome” is dose limiting and includes leukocytosis, fever, dyspnea, chest pain, tachycardia, hypoxia, and sometimes death; corticosteroids seem to benefit this syndrome; QT prolongation is common; common adverse effects include rash, pruritus, headache, arthralgias, anxiety, bleeding, nausea, and vomiting; liver and renal toxicity are uncommon
Relapsed acute promyelocytic leukemia
0.15 mg/kg/day in 100-250 mL of D5W until remission, not to exceed 60 doses, then up to 25 doses over 5 wk for consolidation starting 3 to 6 wk after achievement of remission
l-Asparaginase (Elspar), colaspase
Naturally occurring enzyme derived from Escherichia coli or Erwinia carotovora that cleaves asparagine, an essential amino acid required by rapidly proliferating cells
After IV or IM injection, the drug is metabolized intravascularly by proteolysis; elimination half-life of 8-30 hr
Hypersensitivity can be life- threatening, requiring anaphylaxis precautions and a 2-unit test dose; coagulopathy is common and requires monitoring; nausea, vomiting, abdominal cramps, anorexia, elevated liver function tests, and transient renal insufficiency are common; lethargy, somnolence, fatigue, depression, and confusion are seen, as are pancreatitis and fever
ALL; also used in AML, late-stage CML, CLL, and non-Hodgkin lymphomas
After a 2-unit intradermal test dose, an IM dose of 6000-10,000 IU/m2 every 3 days for 9 doses, or 1000 IU/kg/day IV over 30 min for 10 days, has been used
PEG-Asparaginase (Oncaspar)—pegaspargase
Naturally occurring enzyme, covalently linked to polyethylene glycol to reduce immunogenicity, slow metabolism, and prolong half-life; the enzyme cleaves asparagine, an essential amino acid required by rapidly proliferating cells
When given by IM injection, it has an elimination half-life of approximately 5 days; clearance is not dependent on renal or hepatic function
Although less immunogenic that the non-PEGylated form, hypersensitivity and anaphylaxis can still occur; toxicities similar to those of the non-PEGylated forms are seen, including elevated liver enzymes, coagulopathy, hypercholesterolemia, pancreatitis, hyperglycemia, fever, chills, anorexia, lethargy, confusion, headache, seizures, and azotemia
ALL, and like asparaginase, it is also used for other leukemias and non-Hodgkin lymphomas
2,500 IU/m2 IM every 14 days with other chemotherapy agents for induction or maintenance
Asparaginase Erwinia chrysanthemi (Erwinaze)
Substitute for the naturally occurring enzyme
Not characterized
Severe hypersensitivity reactions, including anaphylaxis
ALL
25,000 IU IM 3×/wk for 6 doses per cycle
Axitinib (Inlyta)
Avoid strong CYP3A inhibitors or reduce dose
Decrease dose 50% for CYP3A inhibitors, renal, hepatic impairment
Diarrhea, hypertension, fatigue, nausea
Advanced renal cell carcinoma after one treatment failure
5 mg BID oral
Azacytidine (Vidaza)
Antimetabolite; induces hypomethylation of DNA, either inducing apoptosis or restoring normal function; at higher doses, acts as a cytidine analog
Not orally bioavailable; metabolized by the liver and excreted in urine; elimination half-life of 4 hr
Myelosuppression is dose limiting; leukopenia, thrombocytopenia, and transient elevation of liver function tests are common; nausea and vomiting and abdominal pain are common
Myelodysplastic syndromes
75-100 mg/m2 for 7 days; repeated every 4 wk for 4 to 6 cycles
Bacillus Calmette- Guérin (TICE BCG, TheraCys), BCG
Immunostimulant/vaccine; induces a cellular immune response at the site of instillation
BCG is a live, attenuated bacteria culture; it does not enter the body in viable form; it has no detectable pharmacokinetic fate; in rare cases, a clinical infection can result from treatment, indicating invasion of the body at site of administration into systemic circulation
Urinary symptoms predominate, including dysuria, hematuria, hesitancy, urgency, frequency, and secondary infection; other toxicities include fever, chills, malaise, myalgias/arthralgias, anorexia, nausea, vomiting, and anemia; clinical mycobacterial infection is rare and generally seen only in immunocompromised patients
Intravesical instillation for noninvasive bladder cancer after removal of papillary tumors; also used for some experimental vaccine programs as an adjuvant to the vaccine
81 mg per treatment, in 53 mL total volume, instructions as previously given; given once weekly for 6 doses and then at 3, 6, 12, 18, and 24 mo after the induction
Bendamustine (Treanda)
Alkylating agent
Reduce dose for hematologic toxicity
Nausea, pyrexia, vomiting, hematologic abnormalities
CLL and B-cell non-Hodgkin lymphoma
CLL: 100 mg/m2 over 60 min, day 1, 2 of 28-day cycle; B-cell: 120 mg/m2 over 30 min on day 1, 2 of 21-day cycle
Bevacizumab (Avastin)
Recombinant humanized monoclonal antibody that binds to all forms of VEGF, preventing binding to its receptors
Administered by IV infusion; half-life is 20 days; the fate of parent drug and metabolites is unknown
Asthenia, pain, nausea/vomiting, diarrhea, anorexia, stomatitis, dermatitis, hypertension, proteinuria; infusion-related reactions rare; hemoptysis, hemorrhage, delayed wound healing, GI perforations; increased risk of thromboembolic events can be severe or fatal
Metastatic colorectal cancer and NSCLC; renal cell carcinoma
5-15 mg/m2/day IV every 3 wk
Bexarotene (Targretin)
Synthetic retinoid, differentiating agent
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access