Fig. 11.1
Incidence of ovarian cancer, United States SEER 18 regions, 2000–2011, by histologic type
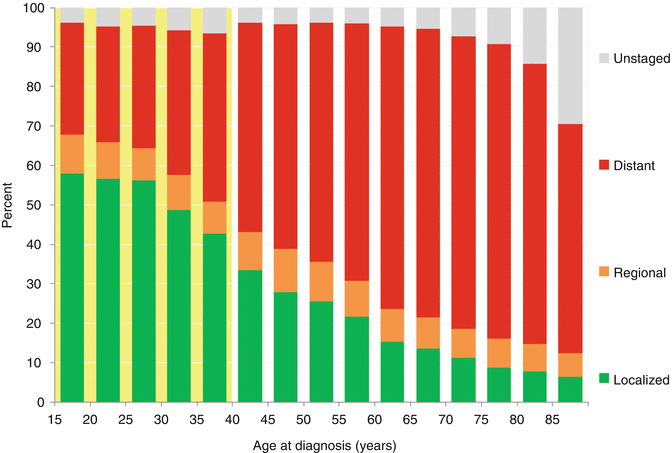
Fig. 11.2
Distribution of ovarian cancer, United States SEER 18 regions, 2000–2011, by histologic type and age
In women with ovarian cancer over 15 years of age, the younger the patient, the more likely the disease is to be localized and therefore of earlier stage (Fig. 11.3). This may reflect the tendency for germ cell tumors to be present in younger females (Fig. 11.2), as these tumors tend to be detected at an earlier stage. As the disease type shifts away from germ cell tumors with increasing age, distant disease at diagnosis increases with age. By age 40, a woman is equally likely to have distant metastases at diagnosis as localized disease. As a result of young women having a favorable histologic subtype (GCTs) with early stage at diagnosis, the prognosis for AYA women with ovarian cancer is better than that for older woman with ovarian cancer. This is unlike most other types of cancer, in which older patients fare better than AYA patients.
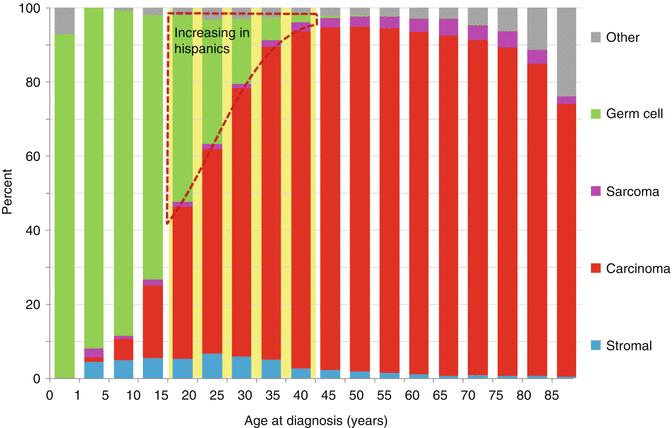
Fig. 11.3
Distribution of ovarian cancer by stage at diagnosis, United States SEER 18 regions, 2000–2011
Figure 11.4 depicts the incidence of ovarian cancer in the United States by age and race/ethnicity. In AYAs (inset), all major races/ethnicities except native North Americans have similar incidence rates as a function of age. The incidence of ovarian cancer in native North Americans was one-third to one-half of any other racial/ethnic group. Native North Americans had one-third to one-half of the incidence. In older women, non-Hispanic whites had the greatest incidence and Asians/Pacific Islander had the lowest incidence.
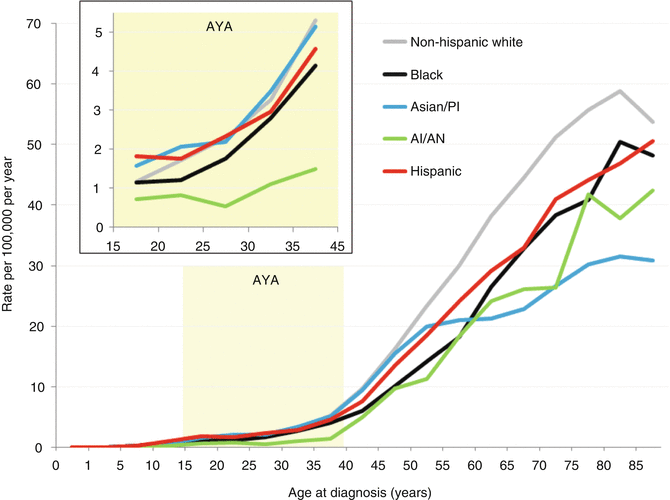
Fig. 11.4
Incidence of ovarian cancer, United States SEER 18 regions, 2000–2011, by age and race/ethnicity
11.2.1.2 Incidence Trends
The incidence of ovarian cancer has been largely unchanged in AYA females in the United States since 1996 [4]. However, the incidence of GCT has increased slightly in premenopausal Asian women and substantially in premenopausal Hispanic women since 1973. In 20- to 44-year old Hispanic women, whose incidence can only be tracked in SEER since 1992, the diagnosis of ovarian GCTs is dramatically increasing, especially since the late 1990s (Fig. 11.5). No other age or ethnic group demonstrates this trend. Potential explanations for this increase include improved reporting, disparate use of oral contraceptives, increasing obesity in this demographic, and delay of childbirth to an older age.
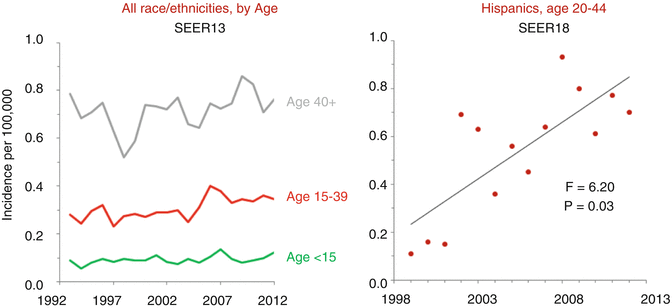
Fig. 11.5
Annual incidence of ovarian GCTs, United States SEER 9 regions, 1975–2011, by age
11.2.2 Pathology and Biology
There are multiple tissue types present in the normal ovary, including germ cells, stroma, and surface epithelium (Tables 11.1 and 11.2). Table 11.2 lists all the histologic types reported in at least ten AYA females (age 15–39) during 2000–2011 in the SEER 18 regions of the United States. Each of these can give rise to a set of distinctive tumors that can occur in pure or combined forms. The majority of ovarian malignancies involve germ cells. Epithelial and stromal tumors become more prevalent throughout the adolescent and young adult years.
Table 11.1
World Health Organization classification of tumors
I. Germ cell tumors (GCT) |
A. Primitive GCT |
1. Dysgerminoma |
2. Endodermal sinus tumor (yolk sac tumor) |
3. Embryonal carcinoma |
4. Polyembryoma |
5. Nongestational choriocarcinoma |
6. Mixed GCT |
B. Biphasic or triphasic teratoma |
1. Immature teratoma (grades 1–3) |
2. Mature teratoma |
3. Solid |
4. Cystic (dermoid) |
5. Fetiform teratoma |
C. Monodermal teratoma and somatic-type tumors |
1. Thyroid (struma ovarii) |
2. Carcinoid |
3. Neuroectodermal |
4. Carcinoma |
5. Melanocyte |
6. Sarcoma |
7. Sebaceous |
8. Pituitary type |
9. Other |
II. Sex cord-stromal tumors |
A. Granulosa cell tumors (adult, juvenile) |
B. Sertoli-Leydig cell tumors |
C. Gynandroblastoma |
D. Sex cord tumor with annular tubules (SCTAT) |
E. Pure stromal tumors (fibroma, thecoma, fibrosarcoma) |
F. Other stromal tumors (sclerosing stromal tumor, Signet ring stromal tumor) |
G. Steroid cell tumors (stromal luteoma; Leydig cell tumor; steroid cell tumor, not otherwise specified) |
III. Epithelial ovarian cancers |
A. Serous |
B. Mucinous |
C. Clear cell |
D. Endometrioid |
E. Brenner |
F. Mixed |
G. Undifferentiated |
Table 11.2
Incidence of ovarian cancer by histologic type, 2000–2011 SEER18 with ≥10 cases in AYAs of age 15–39 years
Histology | Incidence per 100,000, age adjusted | Number in SEER18 | ||||
|---|---|---|---|---|---|---|
Age (years): | <15 | 15–39 | 40+ | <15 | 15–39 | 40+ |
SEER population (×106): | 102.5 | 174.0 | 226.6 | |||
8,460/3: papillary serous cystadenocarcinoma | 0.35 | 6.33 | 2 | 581 | 14,531 | |
8,380/3: endometrioid carcinoma | 0.34 | 2.61 | 0 | 573 | 5,961 | |
9,080/3: teratoma, malignant, NOS | 0.13 | 0.26 | 0.03 | 137 | 450 | 57 |
9,060/3: dysgerminoma | 0.07 | 0.22 | 0.02 | 70 | 389 | 32 |
8,480/3: mucinous adenocarcinoma | 0.20 | 0.83 | 3 | 342 | 1,913 | |
8,441/3: serous cystadenocarcinoma, NOS | 0.17 | 3.92 | 1 | 289 | 9,009 | |
8,470/3: mucinous cystadenocarcinoma, NOS | 0.13 | 0.42 | 4 | 229 | 970 | |
8,310/3: clear cell adenocarcinoma, NOS | 0.11 | 1.29 | 0 | 187 | 2,983 | |
8,620/3: granulosa cell tumor, malignant | 0.10 | 0.3 | 4 | 176 | 684 | |
9,071/3: yolk sac tumor | 0.05 | 0.09 | 0.01 | 48 | 166 | 15 |
8,010/3: carcinoma, NOS | 0.09 | 1.86 | 0 | 160 | 4,444 | |
8,140/3: adenocarcinoma, NOS | 0.09 | 3.59 | 0 | 146 | 8,379 | |
8,462/1: serous papillary cystic tumor, borderline malig. | 0.07 | 0.12 | 0 | 127 | 257 | |
9,085/3: mixed germ cell tumor | 0.06 | 0.07 | 0.01 | 57 | 122 | 14 |
8,000/3: neoplasm, malignant | 0.06 | 1.44 | 1 | 109 | 3,539 | |
8,472/1: mucinous cystic tumor of borderline malig. | 0.06 | 0.12 | 0 | 109 | 262 | |
8,323/3: mixed cell adenocarcinoma | 0.06 | 0.88 | 0 | 106 | 2,010 | |
8,461/3: serous surface papillary carcinoma | 0.06 | 1.20 | 0 | 103 | 2,749 | |
8,010/2: carcinoma in situ, NOS | 0.05 | 0.09 | 2 | 88 | 207 | |
8,442/1: serous cystadenoma, borderline malignancy | 0.04 | 0.08 | 0 | 72 | 170 | |
8,041/3: small cell carcinoma, NOS | 0.01 | 0.04 | 0.04 | 6 | 64 | 81 |
8,240/3: carcinoid tumor, malignant | 0.04 | 0.09 | 1 | 61 | 197 | |
8,631/3: Sertoli-Leydig cell tumor, poorly differentiated | 0.01 | 0.02 | 0.02 | 7 | 36 | 45 |
8,260/3: papillary adenocarcinoma, NOS | 0.02 | 0.55 | 0 | 28 | 1,271 | |
8,070/3: squamous cell carcinoma, NOS | 0.01 | 0.07 | 0 | 25 | 168 | |
8,440/3: cystadenocarcinoma, NOS | 0.01 | 0.15 | 0 | 24 | 354 | |
8,471/3: papillary mucinous cystadenocarcinoma | 0.01 | 0.05 | 0 | 24 | 125 | |
8,634/3: Sertoli-Leydig, poor diff. w. heterologous | 0.01 | 0 | 2 | 22 | 9 | |
9,090/3: struma ovarii, malignant | 0.01 | 0.02 | 0 | 22 | 34 | |
8,950/3: Mullerian mixed tumor | 0.01 | 0.38 | 0 | 20 | 876 | |
9,064/3: germinoma | 0.01 | 0.01 | 0 | 11 | 18 | 9 |
8,560/3: adenosquamous carcinoma | 0.01 | 0.04 | 0 | 16 | 101 | |
8,020/3: carcinoma, undifferentiated type, NOS | 0.01 | 0.11 | 0 | 15 | 242 | |
8,050/3: papillary carcinoma, NOS | 0.01 | 0.15 | 0 | 12 | 352 | |
8,255/3: adenocarcinoma with mixed subtypes | 0.01 | 0.09 | 0 | 12 | 201 | |
9,084/3: teratoma with malignant transformation | 0.01 | 0.01 | 0 | 12 | 34 | |
8,450/3: papillary cystadenocarcinoma, NOS | 0.01 | 0.07 | 0 | 11 | 155 | |
8,590/3: sex cord-gonadal stromal tumor, malig., NOS | 0.01 | 0.01 | 1 | 11 | 20 | |
9,081/3: teratocarcinoma | 0.01 | 0 | 1 | 11 | 2 | |
9,100/3: choriocarcinoma | 0.01 | 0 | 2 | 11 | 7 | |
8,246/3: neuroendocrine carcinoma | 0.01 | 0.04 | 0 | 10 | 101 | |
8,470/2: mucinous cystadenocarcinoma, noninvasive | 0.01 | 0.01 | 0 | 10 | 14 | |
8,980/3: carcinosarcoma, NOS | 0.01 | 0.30 | 0 | 10 | 700 | |
11.2.2.1 Germ Cell Tumors
Germ cell tumors (GCTs) arise from germ cells present in the normal ovary. Benign GCTs are almost always teratomas. Malignant GCTs include dysgerminoma, immature teratoma, yolk sac tumors, embryonal carcinoma, polyembryoma, and nongestational choriocarcinoma and are derived from one or more types of primitive cells or malignant adult components of teratomas [9]. They often display heterogeneous histologic differentiation with multiple histologic subtypes within the same tumor mass. Based on trial enrollment in the Pediatric Intergroup, 60–70 % of patients present with stage I disease, while 25–30 % present with advanced-stage disease. The stage distribution of malignant ovarian GCT in girls older than 12 years was 36 % stage I, 8 % stage II, 46 % stage III, and 10 % stage IV. Most early-stage tumors are mixed GCTs, comprised of immature teratomas with elements of yolk sac tumor. Among 74 high-risk patients with stage III–IV disease, the most common histologies were yolk sac tumors (38.9 %), dysgerminomas (23.6 %), and mixed GCTs (23.6 %) [10, 11]. Upon gross inspection, most malignant ovarian GCTs are unilateral and large, with a median size of 16 cm, ranging from 7 to 40 cm [8].
Historically, a 3-grade system has been used to characterize immature teratomas, but a two-tier system has been proposed due to interobserver and intraobserver inconsistency with the 3-grade system [12]. Tumors should be sampled well (one block for each centimeter of the largest tumor diameter).
Investigators have identified molecular changes specific to GCTs, but clinical application is premature. Overexpression of specific microRNAs (miR 371Y373 and miR 302 clusters) appears to be present in all malignant ovarian GCTs and correlate with response [13]. These microRNAs may aid initial diagnosis and have utility in monitoring response to treatment. In addition, advanced dysgerminomas harbor KIT mutations, which may be a targetable mutation in some patients with advanced metastatic or recurrent disease [14].
Ovarian GCTs bear many histologic and biologic similarities to testicular GCTs (seminomas). However, testicular GCTs in males tend to arise several years after the development of puberty, while ovarian GCTs in females can occur anytime after birth and are much more common in preadolescents. Genetic analysis of ovarian GCTs that present in the second decade of life reveals isochromosome 12p, the characteristic cytogenetic abnormality found in testicular GCT [15–17]. Biologic studies from early cooperative pediatric GCT trials showed that such cytogenetic aberrations were age dependent. Chromosome I(12p) abnormality has been reported in tumors from pubertal and postpubertal males, but the most common abnormalities in prepubertal females in order of prevalence were gains of 1q, +14, +8, +12, +2, +3, and +7 [18].
Comparative genomic hybridization (CGH) has demonstrated recurrent deletions of 6q and 1p in childhood yolk sac tumors [19]. The most commonly detected region of loss was 6q25-6qter, a finding which is noted in several other human tumors, including ovarian, breast, and hepatocellular carcinoma. Multipotent imprinting analysis showed that gonadal and nongonadal GCTs are derived from primordial germ cells that have lost imprinting of small nuclear ribonucleoprotein N (SNRPN) gene and partial loss of H19 and IGF2 [20]. Cooperative pediatric GCT trials from Pediatric Oncology Group (POG)/Children’s Cancer Group (CCG) and Maligne Keimzelltumoren (MAKEI) found that all pure teratomas had normal CGH patterns [21]. Although there were few ovarian GCT specimens in these studies, monoallelic expression of H19 and IGF2 was seen, suggesting no loss of imprinting. Further studies are needed in adolescents and young adults with ovarian GCTs to identify genes that are important to pathogenesis and therefore potential targets for future treatments.
11.2.2.2 Sex Cord-Stromal Tumors
Sex cord-stromal tumors originate from the specialized gonadal stromal cells and their precursors. Granulosa cells and Sertoli cells arise from sex cord cells, while theca cells, Leydig cells, lipid cells, and fibroblasts arise from stromal cells and their pluripotent mesenchymal precursors. These tumors can occur as an isolated histologic type orin combination, and together they account for 7 % of all ovarian malignancies [22] and approximately 5 % of ovarian malignancies in women ages 15–24 years [5]. Granulosa cell tumors are the most common subtype and have both adult and juvenile subtypes. Juvenile granulosa cell tumors are more often found in the AYA population, but adult-type tumors can be found in this age range as well; diagnosis is made by histologic criteria and not patient age [23].
Gross inspection usually reveals a unilateral solid adnexal mass, often yellow and multilobulated in appearance or hemorrhagic with hemoperitoneum. Definitive diagnosis can be difficult on frozen section, and immunohistochemical staining may ultimately be useful, as inhibin, calretinin, and FOXL2 may be expressed. A reticulin stain may also be positive in patients with adult granulosa cell tumor and help differentiate it from fibrothecoma.
Sertoli-Leydig cell tumors recapitulate the phases of testicular development and may act in an aggressive fashion. It is therefore essential to distinguish it from the benign Sertoli cell tumor, which consists only of Sertoli cells. These tumors are categorized into three forms: well-differentiated, intermediate differentiation (based on immature Sertoli cells), and poorly differentiated forms (sarcomatoid or retiform). Over 95 % are unilateral.
A subgroup of the sex cord tumor grouping is the ovarian sex cord tumor with annular tubules. The biologic behavior of this lesion is thought to be intermediate between granulosa cell tumors and Sertoli cell tumors and is associated with Peutz-Jeghers syndrome [24, 25]. Patients should be carefully screened for adenoma malignum of the cervix, as 15 % of patients harbor an occult lesion.
Significant progress has been made in the molecular biology of stromal ovarian tumors. A missense point mutation in the FOXL2 gene (a 402C to G mutation) has been found in nearly all adult granulosa cell tumors and has been absent in other pure subtypes of stromal tumors [26]. This may be useful in diagnosis and may present an opportunity for targeted therapy in the future.
The presence of DICER1 mutations in stromal tumors, especially Sertoli-Leydig cell tumors, marks a major discovery important in diagnosis and detection of a previously unrecognized familial syndrome. Schultz et al. analyzed kindreds of 325 children with pleuropulmonary blastoma (PPB), a childhood syndrome that is usually fatal unless detected early. Among this cohort, two of the three children with both PPB and Sertoli-Leydig cell tumors had germline DICER1 mutations. In addition, six family members had stromal ovarian cancers, and four of these six patients had germline DICER1 mutations [27]. Subsequently, 14 of 26 patients (60 %) with Sertoli-Leydig cell tumors had somatic DICER1 mutations; four also had germline DICER1 mutations [28]. Schultz et al. have founded an Ovarian Stromal Tumor Registry in order to better characterize these tumors and identify babies at risk for PPB as well as Sertoli-Leydig cell tumors.
11.2.2.3 Epithelial Carcinomas
The primary subtypes of epithelial carcinoma in young women are the serous and mucinous types, but BRCA-associated serous cancers, low malignant potential tumors, and clear cell carcinomas are also found in the AYA population [29]. Adenocarcinoma is found very rarely before the age of 24 years [5]. Little information is available about the biologic issues unique to young women with epithelial ovarian cancer. However, approximately 10 % of ovarian cancers are caused by mutations in the tumor suppressor genes BRCA1 and BRCA2, and high-grade serous tumors with BRCA1 mutations tend to occur in women at younger ages than non-BRCA-mutated cancers. Females with a personal history of ovarian cancer, breast and ovarian cancer, and breast cancer at age 50 or younger with a close relative with ovarian cancer or a close relative with male breast cancer at any age; women of Ashkenazi Jewish ancestry in whom breast cancer was diagnosed at age 40 years or younger; or women with a close relative with a known BRCA mutation should be tested, and family members should be offered testing should the results of the index patient be positive. Not only is BRCA testing useful for confirmation of pathology, but AYA patients with a BRCA mutation require specific counseling regarding medical and surgical risk reduction [30].
Low-grade serous ovarian carcinomas are also found in women at a younger age than the typical high-grade serous ovarian counterpart. A two-tier grading system has been proposed to improve consistency in diagnosis. In this system, low-grade serous tumors have mild to moderate nuclear atypia and a mitotic index of up to 12 mitoses/10 high-power fields [31]. Analysis of clustering supports this approach, as high-grade serous ovarian cancers have a profile distinct from low-grade and borderline tumors, which resemble each other. Compared with high-grade serous ovarian cancer, low-grade serous ovarian cancers are more likely to express estrogen/progesterone receptors and e-cadherin and less likely to express p53, bcl2, WT1, HER-2/neu, c-KIT, Ki-67, or MMP-9 [32, 33].
Clear cell carcinomas may also occur in the AYA patient, and these tumors show a preponderance of ARID1A mutations.
11.2.2.4 Tumors of Low Malignant Potential
Tumors of low malignant potential (LMP) represent a category of neoplasms that are distinct from benign cystadenomas and cystadenocarcinomas. First described by Taylor in 1929, they have since been referred to as borderline tumors or atypically proliferating tumors [34, 35]. These tumors arise from the surface epithelium of the ovary, and 80–95 % are of serous or mucinous histology. LMP tumors of the ovary comprise approximately 15 % of all epithelial ovarian tumors, and 71 % occur in premenopausal women at a median of 40 years [36–38]. The pathologic criteria for diagnosis of these tumors include the absence of stromal invasion in the ovary and any two of the following characteristics: epithelial “tufting,” multilayering of the epithelium, mitotic activity, and nuclear atypia. Trisomy 12 has been reported in LMP tumors [39].
11.2.3 Presentation and Evaluation
Despite their low incidence, ovarian tumors represent a major diagnostic and treatment dilemma for treating physicians. While optimal care is provided through a multidisciplinary approach incorporating pediatric oncologists, pediatric surgeons, and gynecologic oncologists, this is often not the case. The rare nature of these diseases leads to lack of awareness by some treating physicians and may result in unnecessary second surgeries, otherwise unnecessary adjuvant therapy, and failure to preserve fertility and lack of age-appropriate care. Adolescents with ovarian tumors are grossly underrepresented on clinical trials, whether pediatric or adult cooperative group in origin, a finding which may adversely affect survival [40].
Appropriate treatment of ovarian tumors is determined by many factors, including patient age, karyotype, extent of disease, tumor histology, and comorbid conditions. Conservative surgery to maintain reproductive potential is an important consideration in all adolescents and young adults and is usually feasible. Appropriate surgical staging and assessment are necessary components in determining the extent of surgery required and the need for postoperative chemotherapy. An appropriate initial evaluation should be used to tailor therapy.
The most common presenting signs and symptoms of an ovarian tumor are abdominal pain, palpable abdominal mass, increasing abdominal girth, urinary frequency, constipation, and dysuria [41, 42]. Some tumors are asymptomatic and are discovered during routine examination. Abdominal pain is most often chronic, but torsion of an enlarged ovary can result in acute pain. Since normal stromal cells produce steroid hormones, physical manifestations of excess estrogen or androgen production (early menarche, hirsutism, virilism) suggest the possibility of a sex cord-stromal tumor. Gynandroblastomas, while rare, are composed of granulosa cells, tubules, and Leydig cells and can cause premature breast development, hyperestrogenism, or androgenism in adolescents [24, 25]. Isosexual precocity may also be seen in mixed malignant GCTs due to tumor production of β-HCG [43]. A complete history and physical examination should be performed, including abdominal palpation. Pelvic and rectal examination by a skilled practitioner should be considered and performed when appropriate; this may be delayed to an examination under anesthesia in the very young.
A panel of laboratory values should be obtained, including αFP, β-HCG, lactate dehydrogenase (LDH), CA-125, carcinoembryonic antigen (CEA), estradiol, testosterone, F9 embryoglycan, inhibin A and B, and anti-Mullerian hormone (AMH) [39, 44]. Elevated alpha fetoprotein (αFP) usually indicates a component of yolk sac tumor. An elevation of β-human choriogonadotropin (β-HCG) is always seen in choriocarcinoma but may also be present in embryonal carcinoma, polyembryoma, pure ovarian dysgerminoma, and mixed germ cell tumors [41, 44] (Table 11.3).
Imaging studies may include a pelvic ultrasound to delineate the characteristics of the pelvic organs, specifically the ovaries. Specific characteristics may suggest the diagnosis, such as the presence of teeth in teratomas. A computed tomographic (CT) scan of the abdomen and pelvis may be helpful to determine the extent of disease preoperatively. If the ovarian mass is complex or solid, over 8 cm, and has persisted for more than 2 months, or if there is evidence of extraovarian disease, surgical exploration is indicated [45].
Gonadal dysgenesis, a developmental anomaly in which sex steroid production is diminished, is a risk factor for malignant ovarian GCTs. Patients may present with delayed puberty or primary amenorrhea. Malignant ovarian GCTs, especially dysgerminomas, can develop in up to 30 % of patients with Swyer syndrome (complete gonadal dysgenesis, with a 46 XY genotype and a female phenotype). Prophylactic removal of both gonads is indicated upon diagnosis of this syndrome [46]. Gonadoblastoma can develop in patients with Turner syndrome when mosaicism includes a Y chromosome [47]. These women should be screened with yearly transvaginal ultrasound and CA-125 beginning at age 25–30 years, and they should consider prophylactic oophorectomy after completion of childbearing or at the age of 35 years [48].
Based on this information, the evaluation for a young patient with an adnexal mass suspicious for malignancy should include routine preoperative blood work, chest radiograph, serum tumor markers guided by symptoms and signs, pelvic/transvaginal ultrasonography, and CT of the abdomen/pelvis. Indications for surgery in a child with an ovarian mass include persistent pain, suspicion of torsion, hydronephrosis, complex or solid mass on imaging, metastasis, ascites, positive tumor markers, unclear origin of mass, persistence or growth of cyst on serial imaging, large masses with complex features, and rapid virilization, estrogenization, or precocious puberty [49].
11.2.4 Surgical Treatment Guidelines
11.2.4.1 Surgical Management
When an adolescent or young adult patient presents with an adnexal mass, precise histology is often difficult to determine during intraoperative pathology consultation. Gross inspection does not yield a diagnosis, and recommendations for adjuvant therapy can only be based on final pathology with complete staging information. Therefore, it is imperative that the surgeon obtains the necessary information to determine the diagnosis and need for further therapy while preserving fertility in these young patients. The goals of surgery are to obtain the diagnosis, perform comprehensive surgical staging in clinically apparent early disease, and perform maximal cytoreductive surgery in patients with advanced disease. Often, this must be done in the setting of unclear intraoperative pathology, and the surgeon must then rely on preoperative imaging and tumor markers to gauge the extent of surgery [49]. The balance is obtaining enough appropriate samples to guide therapy, treat the disease surgically, and preserve reproductive function while avoiding the need for re-exploration. Immediate consultation with a gynecologic oncologist, if not done preoperatively, should occur intraoperatively at the time of diagnosis.
Fertility-Sparing Surgery
Young women who present with ovarian masses generally have many concerns regarding future reproductive potential. Preoperative discussions should occur to review options for maintaining ovarian and/or uterine function based on potential operative findings. In general, the unilateral nature of most of these tumors, preponderance of early-stage disease, and effective chemotherapy have allowed the successful use of conservative, fertility-sparing surgery in most adolescent and young adult patients with limited disease [45, 50]. An early report of 182 patients with early-stage ovarian germ cell tumors revealed no decrease in survival when fertility-sparing surgery was performed [51]. Since that time, fertility-sparing surgery has become the standard of care for AYA patients with limited disease, and it is now reported in the majority of patients [52–54]. While one report has focused on ovarian cystectomy [55], the standard of care refers to unilateral salpingo-oophorectomy with preservation of a normal-appearing contralateral adnexa and uterus [45]. Due to the low yield from random ovarian biopsy, and the potential for disruption of reproductive potential due to adhesions or trauma, the routine biopsy of a normal-appearing contralateral ovary is not advised [56]. These recommendations hold true for patients with germ cell tumors, sex cord-stromal tumors, and epithelial carcinomas [25, 57]. Consideration should be given to removal of the residual ovary once childbearing is complete [29].
One caveat to fertility-sparing surgery is the diagnosis of dysgenetic gonads, which must prompt bilateral salpingo-oophorectomy. This finding is rarely associated with dysgerminoma, in which a dysgenetic gonad is present in a phenotypically normal female with abnormal karyotype. The contralateral dysgenetic or “streak” gonad also carries a high potential for a future malignant GCT. Therefore, in cases of intraoperative diagnosis of dysgerminoma, the pathologist should be asked to carefully evaluate any residual normal ovary and look for any elements of gonadoblastoma. As the pathologist is evaluating the specimen further, the surgeon should inspect the contralateral adnexa to determine whether a normal ovary or streak gonad is present. Normal ovarian tissue excludes the possibility of dysgenetic gonads, thereby allowing the surgeon to conserve the contralateral ovary and preserve reproductive potential. However, in the event of a “streak” gonad or diagnosis of gonadoblastoma, a bilateral salpingo-oophorectomy should be performed to remove any gonadal tissue, regardless of age [45, 58].
Even in the rare circumstance where both ovaries must be removed, the uterus should be left in place to allow for future assisted reproductive techniques using donor oocytes [56, 59, 60]. The only absolute indication for uterine removal in this group of patients is adenoma malignum of the cervix in patients with Peutz-Jeghers syndrome associated with ovarian sex-cord tumor with annular tubules [24, 25].
When an adnexal mass is found to be a mature cystic teratoma, areas of squamous differentiation and small nodules in the wall of the cyst should be evaluated for malignant elements or for immature neural tissue. These indicate malignancy and should prompt surgical staging. If no malignant elements are identified, the neoplasm is benign and an ovarian cystectomy is sufficient. The contralateral ovary should be inspected, as 12 % of cases are bilateral. If a cyst is found in the contralateral ovary, a cystectomy should be performed, preserving as much normal ovarian tissue as possible. The cyst should be sent for immediate histologic evaluation, and if malignant disease is revealed, bilateral oophorectomy is performed. In 5–10 % of malignant GCTs, there is an associated contralateral benign mature cystic teratoma, and in these situations, the remainder of the benign ovary can be preserved [61].
Patients with large ovarian cysts where the cortex has become quite thin still benefit from cystectomy with preservation of as much healthy cortex as possible, as this continues to function postoperatively [49].
Comprehensive Surgical Staging
The majority of AYA patients with a malignant adnexal mass present with clinically early-stage disease. The patient with no gross extraovarian disease should undergo comprehensive surgical staging in order to determine the stage, need for adjuvant therapy, and prognosis, as up to 30 % of patients can have occult disease. Staging consists of cytologic washings, infracolic omentectomy, and peritoneal biopsy specimens from each paracolic gutter, the vesicouterine fold, and the pouch of Douglas. The bowel should be inspected from the ileocecal valve to the ligament of Treitz, specifically evaluating for tumor implants and sites of obstruction. All peritoneal surfaces should be carefully inspected. Any suspicious areas should be sampled. Pelvic and para-aortic lymph node sampling is recommended for full staging, except in patients with sex cord-stromal and mucinous carcinomas [62, 63].
A different approach has been espoused in pediatric patients with ovarian germ cell tumors. A review of deviations from standard surgical guidelines failed to show any impact on survival. Therefore, a proposal for more limited surgical staging includes collection of ascites or cytologic washings, examination of peritoneal surfaces with biopsy or excision of any nodules but no random biopsies of normal-appearing peritoneum, examination and palpation of retroperitoneal lymph nodes and sampling only of any firm or enlarged nodes, inspection and palpation of omentum with removal only of any adherent or abnormal areas, biopsy of any abnormal areas, and complete resection of the tumor-containing ovary with sparing of the fallopian tube if uninvolved [56]. This approach omits random biopsies of peritoneum, systemic lymphadenectomy, omentectomy, and salpingectomy and targets only tissue that is abnormal upon inspection. This has been suggested only for pediatric germ cell tumors, and prospective evaluation is in the planning stage.
Patients with malignancies who have not been staged at the time of their initial surgery present a dilemma – should reoperation proceed solely for the purpose of comprehensive surgical staging? For patients with apparent early invasive epithelial cancers (e.g., mucinous and clear cell carcinomas), comprehensive surgical staging is the standard approach, as the recommended treatment may drastically change with upstaging and outcomes in the setting of recurrent disease are dismal. In patients with apparent early-stage borderline tumors, germ cell tumors, and sex cord-stromal tumors, however, one approach is to obtain imaging studies and, if negative, omit a second procedure for surgical staging. This approach is supported in GCTs by the pediatric intergroup study, as noted above, and by a recent report from the Multicenter Italian Trials in Ovarian Cancer (MITO) group, in which 21/26 patients with clinically apparent early-stage pure ovarian dysgerminoma were unstaged at primary surgery. No additional surgery was performed and no adjuvant chemotherapy was given; three patients relapsed and all were cured [64]. Patients with evidence of limited extraovarian GCT who did not have complete staging do not usually warrant repeat surgery, as chemotherapy is indicated.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


