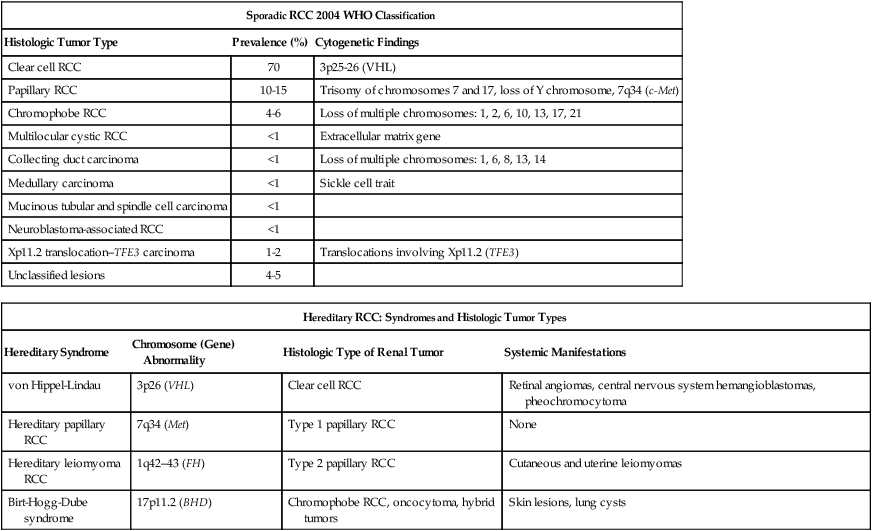
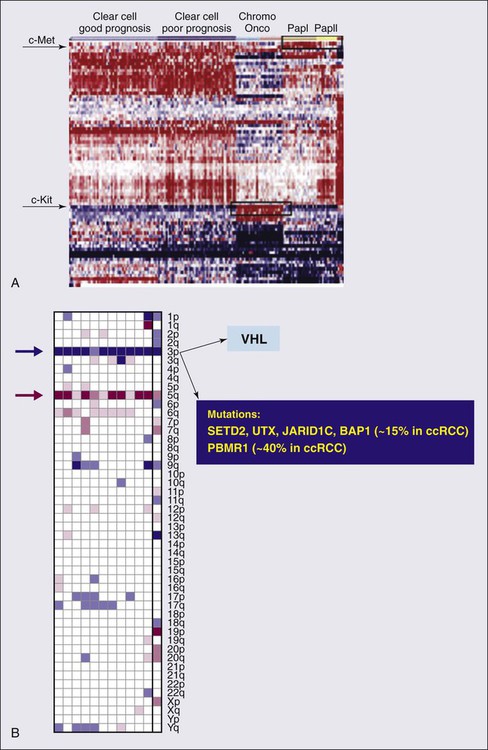
82 Roberto Pili, Eric Kauffman and Ronald Rodriguez • Renal cell carcinoma (RCC) accounts for 3% of malignancies in adults. • Cigarette smoking (in more than 20% of cases) and obesity (in more than 30%) are established causal factors for RCC. • Four percent of cases of RCC arise from hereditary syndromes. • Different subtypes of RCC are characterized by distinct clinical behavior, genetic abnormalities, and molecular signatures. • Clear cell RCC is the most common histologic subtype, representing approximately 70% of all sporadic RCCs. • The von Hippel-Lindau tumor suppressor gene is genetically and epigenetically altered in more than 75% of sporadic cases of clear cell RCC. • Prognosis for RCC is dependent on tumor histologic type, grade, and stage. • Nephron-sparing surgery has become the gold standard, when feasible. • Follow-up guidelines for resected RCC include history, physical examination, periodic metabolic panels, and abdominal and chest computed tomography (CT) studies 4 to 6 months after surgery. • High-dose bolus interleukin-2 (IL-2), though toxic and of limited use in selected patients, remains a therapeutic option for clear cell RCC because of its potential for durable complete response. Additional immunotherapeutic approaches under development and promising results have been reported with the immunocheckpoint inhibitor PD-1 antibody. Identification of predictors of response to immunotherapies is undergoing. • Antiangiogenesis drugs have become the new standard of care in the first-line setting for clear cell RCC. Clinical benefit has also been shown with vascular endothelial growth factor and mammalian target of rapamycin inhibitors in subsequent lines of therapies. Novel targets for therapeutic interventions have been identified and are being exploited in clinical testing. • Optimal treatment for non–clear cell RCC remains a challenge because of the genetic differences and little knowledge of the dysregulated molecular biology driving these cancers. Rational preclinical and clinical testing is needed. The estimated new cases of kidney and renal pelvis tumors for 2012 was 64,770.1 A predominance of cases in male patients has been reported, with an estimated 40,250 men developing disease in 2012, compared with 24,520 women. The estimated number of deaths in 2013 is 13,570 for both sexes (8,650 in men and 4,920 in women). The increasing incidence of RCC observed in the past had been attributed to increased detection as a result of the widespread use of imaging modalities such as computed tomography (CT), ultrasonography, and magnetic resonance imaging (MRI).2,3 Although a decrease in the size of diagnosed renal cell tumors over time has been noted, an increasing incidence of large and late-stage RCC has also been observed, and partly accounts for the overall increase in incidence.3 In the United States, increases in incidence have been more rapid among women than among men and among African Americans than among whites, leading to a shift in excess from among whites to among African Americans.4 Cigarette smoking and obesity are the most consistently established causal risk factors, accounting for more than 20% and 30% of renal cell cancers, respectively.4,5 Hypertension, rather than antihypertensive drugs, appears to influence renal cell cancer development, although the mechanism is unknown.4,5 This was the conclusion of a large study of 363,992 Swedish men who received at least one physical examination between 1971 and 1992 and who were followed up until death or until the end of 1995.5 The relative risk for RCC was 1.3 for former smokers and 1.6 for current smokers. The relative risk for renal pelvis cancer was even higher at 1.6 for former smokers and 3.5 for current smokers. With regard to obesity, patients with a body mass index in the highest one-eighth of the cohort had a relative risk of 1.9 when compared with patients in the leanest subgroup. Hypertension was confirmed as a third risk factor for RCC. A study tested whether smoking is associated with mutations in the von Hippel-Lindau tumor suppressor gene (VHL) in 337 cases of sporadic RCC among 120,852 people over a mean follow-up period of 11.3 years; the findings suggest that smoking causes RCC independently of VHL mutations.6 Patients with end-stage renal disease have an increased incidence of RCC when compared with the general population. Patients receiving prolonged dialysis tend to develop acquired renal cystic disease, possibly as a result of disordered proliferation within the native kidney. In these patients, the tumors often are bilateral and multifocal, with a papillary histology.7 Accordingly, these patients should be monitored regularly with renal ultrasound examinations or noncontrast MRI. If the patient is on dialysis, then nephrectomy is typically preferred, even when the tumor is smaller than 4 cm, providing the risk of surgery is reasonable. Additional evidences suggest a potential role in RCC for alcohol consumption, occupational exposure to trichloroethylene, and high parity among women. However, further research is needed into the potential causal effects of genetic factors and their interaction with environmental exposures. Large studies employing genome-wide scanning technology are in progress to provide novel discoveries in renal carcinogenesis.8 Kidney tumors usually are unilateral but may be bilateral in 2% to 4% of cases.9 These tumors tend to grow into the renal vein and may form a tumor thrombus that extends into the vena cava and even the right atrium. Vascular involvement is present in 4% to 10% of patients at the time of presentation.10 From a pathological and surgical prospective, it is important to distinguish a tumor thrombus form a positive margin at the vascular surface, as a true positive margin (with actual invasion into the wall of the vessel) portends a poor prognosis. RCC is a clinically and pathologically heterogeneous disease.11 The 2004 World Health Organization (WHO) classification for renal neoplasms recognizes several distinct histologic subtypes of RCC (Table 82-1). These subtypes include clear cell RCC, papillary RCC, chromophobe RCC, hereditary cancer syndromes, multilocular cystic RCC, collecting duct carcinoma, medullary carcinoma, mucinous tubular and spindle cell carcinoma, neuroblastoma-associated RCC, Xp11.2 translocation–TFE3 carcinoma, and unclassified lesions.12,13 Clear cell RCC is the most common adult RCC, representing 70% of all RCCs. Papillary type I and type II RCC account for 10% to 15%, chromophobe RCC for 4% to 6%, collecting duct carcinoma for less than 1%, and unclassified lesions for 4% to 5% of RCCs. Tumors may be composed of mixed histologic subtypes, and each subtype may feature high-grade sarcomatoid characteristics. Histologic differentiation of most subtypes of RCC can be accomplished with hematoxylin-and-eosin staining techniques. The conventional histologic pattern is the most common, characterized by large clear cells with abundant cytoplasm. The chromophobe pattern is granular with abundant mitochondria. The papillary or tubulopapillary variant may represent a different type of tumor, because they tend to be smaller with fewer anaplastic features. The most widely used grading system for RCC is the nuclear grading system developed by Fuhrman and colleagues.14 This system assigns a grade from I to IV, based on nuclear size, roundness, and other morphologic features, such as the prominence of nucleoli and the presence or absence of clumped chromatin. Patients with tumors of high Fuhrman grade tend to have poorer clinical outcomes. However, many pathologists omit Fuhrman grade when the apparent aggressiveness of the histology is not related to prognosis (e.g., chromophobe carcinoma, which tends to have a favorable prognosis even when the cellular characteristics appear aggressive). Table 82-1 Histologic Classification of Renal Cell Carcinoma RCC, renal cell carcinoma; WHO, World Health Organization. Adapted from Prasad SR, Humphrey PA, Catena JR, et al. Common and uncommon histologic subtypes of renal cell carcinoma: imaging spectrum with pathologic correlation. RadioGraphics 2006;26:1795–1806. Until recently, RCC was thought to represent a monomorphic disease arising from a probable common precursor cell but with different histologic and clinical manifestations. Genetic characterization based on cytogenetics and molecular biology has established that different subtypes of RCCs are characterized by distinct genetic abnormalities and molecular signatures reflecting the differences in the cell type, biology, and underlying molecular mechanisms.15 Additional tumor metabolic pathways may explain the biological diversity of RCC. A common genetic feature signature of sporadic clear cell RCC is the loss of chromosome 3p, suggesting the presence of one or more RCC tumor suppressor genes at this site. The von Hippel-Lindau tumor suppressor gene (VHL), which resides on chromosome 3p25, is mutated or silenced in greater than 50% of sporadic clear cell RCCs.16,17 Germline VHL mutations give rise to von Hippel-Lindau syndrome, which is characterized by an increased risk of blood vessel tumors (hemangioblastomas), endocrine tumors and RCC. The VHL gene product, pVHL, is the substrate recognition module of an E3 ubiquitin ligase that targets the hypoxia-inducible factor (HIF) α transcription factors (HIF1α, HIF2α, and HIF3α) for destruction in the presence of oxygen. Hypoxic cells, or cells lacking pVHL (“pseudohypoxic”), accumulate high levels of HIF, which activates the transcription of a variety of genes, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF-3) B, and transforming growth factor alpha (TGF-2). Restoration of pVHL function in VHL−/− mutant renal carcinoma cells suppresses their ability to form tumors experimentally by reducing HIFα levels.18,19 Inhibition of HIFα is necessary and sufficient for tumor suppression by pVHL in RCC nude mouse xenograft assays. This provides a rationale for treating VHL−/− RCC with inhibitors of HIFα or its downstream targets. While the HIF1α isoform was initially believed to be more important, increasing literature supports HIF2α as the more important HIFα member in mediating tumorigenesis.18,19 Although most investigation has focused on the role of HIFα isoforms, the pVHL protein also has several other targets in addition to HIFα, postulated by some to contribute to tumorigenesis. Elucidating these targets will lead to further knowledge of how pVHL suppresses tumor growth. Analysis of mutations in exon 3 of the VHL gene may be useful in refining the diagnostic criteria for conventional RCC versus chromophobe RCC with clear cell features.20 A recent work from Dr. Rathmell’s group have generated gene expression microarray data using software that implements iterative unsupervised consensus clustering algorithms to identify the optimal molecular subclasses. A Consensus Cluster analysis identified two distinct subtypes of clear cell RCC within the training set, designated clear cell type A (ccA) and B (ccB). In each subtype the analysis of data defined a small, highly predictive gene set. A validation data set of 177 tumors was analyzed. Tumors designated ccA had improved disease-specific survival compared with ccB (median survival of 8.6 vs. 2.0 years). These preliminary data suggest that a cluster subtype classification is possible in RCC as performed in other diseases such as breast cancer. Prospective clinical studies will be necessary to validate these important findings.21 The genetic studies in familial RCC have led to the identification of specific molecular signatures in non–clear cell histotypes as well, such as Met hyperactivation in papillary type I, fumarate hydratase mutation in papillary type II, and c-Kit overexpression in chromophobe RCC (see “Papillary Types I and II Renal Cell Carcinoma”). Preliminary results from ongoing genetic studies have shown the possibility of clustering different histotypes based, for example, on kinase expression (Fig. 82-1A).22 DNA, histones, and nonhistone proteins are condensed into a highly complex nucleoprotein structure known as chromatin that acts as a prototype for all of the genetic information. Chromatin conformation can be in a heterochromatin form, a highly compact structure, associated primarily with transcriptionally inactive genes, or a euchromatin form, consisting of a more open or relaxed structure, associated with transcriptionally active genes.23 Epigenetic mechanisms mediated by histone deacetylases (HDACs), histone acetyltransferases, and histone methyltransferases play critical roles in various biological and cellular processes, including cell proliferation, angiogenesis, hypoxia-related effects, and cell-cycle regulation. Histones on the N-terminal are altered by acetylation, methylation, phosphorylation, ubiquitination, sumoylation, deamination, or adenosine diphosphate ribosylation.24,25 The different histone residues and their modifications result in either transcriptionally active or repressive marks. For example, histone H3 lysine 4 (designated as H3K4), H3K36, and H3K79 are associated with active marks, whereas H3K9, H3K27, and H4K20 are associated with repressive marks.26 Several studies have recently reported SETD2, a histone methyltransferase, as a tumor suppressor gene in RCC.27,28 An initial study consisting of a large-scale next-generation sequencing of primary RCC genomes identified somatic truncating mutations in SETD2 (approximately 3%).28 These mutations are associated with a decrease in H3K36 trimethylation levels in several clear cell renal carcinoma cell lines, VHL mutations and/or hypoxic phenotype (determined by a panel of hypoxia-related genes) in clinical samples, as well as a chromatin remodeling complex protein PBRM1.29 It is also important to note that SETD2 and PBRM1 are located on the short arm of the human chromosome 3 (3p21.31), in close proximity to the VHL gene and the 3p region is frequently altered in clear cell RCCs30 (see Fig. 82-1B). Although mutations in enzymes governing chromatin remodeling are often found in advanced stage disease, these mutations have not been correlated to survival.31 The H3K27 methyltransferase EZH2 (which is associated with aggressiveness in breast cancer and overexpressed in metastatic prostate cancer)32 is overexpressed in renal tumor patient samples compared with their normal adjacent.33 Conversely, histone demethylases governing H3K27 methylation status UTX and JMJD3 are overexpressed in clear cell RCCs compared with the adjacent nontumor tissue, which corresponds to the decreased H3K27 methylation in clear cell RCC.34 These preliminary evidences suggest that alterations in histone-modifying genes may be associated with RCC and could be exploited for therapeutic interventions. More recently, the BAP1 protein, a nuclear deubiquitinase, has been reported to be inactivated in 15% of clear cell RCCs. BAP1 mediates suppression of cell proliferation, ubiquitinates the histone 2A lysine 119, and its loss sensitizes RCC cells in vitro to genotoxic stress. Initial evidences suggest that BAP1 loss is associated with high tumor grade and poor prognosis. Interestingly, mutations in BAP1 and PBRM1 appear to be mutually exclusive. These preliminary results provide a rationale for future integrated pathological and molecular genetic classification of RCC that will likely lead to subtype specific treatments.35 Intratumor heterogeneity has always been considered a potential major clinical hurdle to develop personalized-medicine strategies that depend on results from single-tumor biopsy samples. A recent report has now shown very elegantly that intratumor heterogeneity is a reality.36 Different techniques, including exome sequencing, chromosome aberration analysis, and ploidy profiling on multiple spatially separated samples obtained from primary renal carcinomas and associated metastatic sites, determined that 63% to 69% of all somatic mutations were not detectable across every tumor region. Mutational intratumor heterogeneity was seen for multiple tumor suppressor genes and chromatin remodeling genes, including SETD2, PTEN, and KDM5C. Gene-expression signatures associated with good and poor prognosis were detected in different regions of the same tumor. These new intriguing findings, combined with the complex genomic landscape, make us rethink the common assumption that a single genomic test might guide therapy. The timing and location of the tumor sample acquisition remain a challenge, but there is still an opportunity to make personalized medicine a goal to achieve in the future for the treatment of cancer, and RCC in particular. As with clear cell RCC, genetic studies in familial RCC have led to the identification of genes responsible for non–clear cell histotypes as well. However, unlike clear cell RCC, gene mutations identified in hereditary non–clear cell RCC are absent in the vast majority of sporadic cases. Activating mutations in the Met oncogene responsible for hereditary papillary RCC (see “Familial Renal Cell Carcinoma”) are found in only approximately 10% of sporadic papillary type I RCC cases.37 The Met tyrosine kinase receptor localizes to the cell membrane where it binds its extracellular ligand, hepatocyte growth factor, triggering intracellular activation of the Akt, Rac, and MAP kinase signaling pathways, promoting cell proliferation and migration. Hyperactivation of Met signaling is believed to promote tumorigenesis by upregulation of these downstream pathways. Both Met and hepatocyte growth factor localize to chromosome 7, which is commonly amplified in sporadic papillary type I RCC. The fumarate hydratase gene encoding a Krebs cycle enzyme and mutated in hereditary papillary type II RCC (as part of hereditary leiomyomatosis and RCC syndrome; see “Familial Renal Cell Carcinoma”) has not been identified in sporadic papillary type II.38 However, increased activity of the NRF2 transcription factor resulting from fumarate hydratase loss in hereditary papillary type II has also been demonstrated in sporadic papillary type II renal cancers.39,40 As with papillary types of RCC, the genetic mutations underlying sporadic chromophobe RCC tumorigenesis remain to be elucidated, and appear to have little mutational overlap with hereditary chromophobe RCC. The folliculin gene mutated in the most common type of hereditary chromophobe RCC (Birt-Hogg-Dubé; see “Familial Renal Cell Carcinoma”) is rarely mutated (0% to 10%) in sporadic chromophobe RCC tumors.41 While PTEN has been implicated in a rarer type of hereditary chromophobe RCC, its mutation is yet to be identified in the sporadic disease.42 Also known as Xp11 translocation kidney cancer, TFE3-fusion RCC represents <1% of all sporadic renal cell cancers. It is the most recently designated histologic subtype of RCC by the WHO. TFE3-fusion RCC occurs in younger patients and is the most common mutation in pediatric RCC tumors. These tumors are clinically aggressive and commonly present with metastasis, particularly to regional lymph nodes. These tumors harbor a pathognomonic fusion between the TFE3 gene of chromosome Xp11.2 and one of a number of possible fusion partners on various chromosomes, most commonly PRCC, ASPRC1, and SFPQ. The TFE3 gene encodes a transcription factor involved in the regulation of many proteins implicated in carcinogenesis, including TGF, Met, Rb, Folliculin, Ets, and E-cadherin. It is believed that the fusion promotes tumorigenesis by causing dysregulated transcriptional TFE3 activity. Immunohistochemical detection of nuclear TFE3 expression is suggestive of the underlying fusion mutation43; however, definitive diagnosis requires genetic confirmation by karyotype, fluorescence in situ hybridization, or polymerase chain reaction (PCR). Rarely, fusions between the related transcription factor gene, TFEB, and the MALAT1/Alpha gene also are found in renal cancers. Less than 30 cases have thus far been reported. The histology of these tumors appears to be distinct from TFE3-fusion tumors. TFEB-fusion cancers similarly occur in younger patients, but in contrast to TFE3-fusion cancers, appear to confer an excellent prognosis. The function of the TFEB transcription factor is unknown, but a central role in lysosome biogenesis and autophagy regulation has been suggested.44 In a small percentage (5%) of cases, RCC is a feature of one of several hereditary syndromes.15,45 Such syndromes are associated with distinct histologic subtypes of RCC, and in each case patients have increased risk of multifocal tumor development.15,45 Management is dependent on preservation of renal function. Close surveillance and minimization of surgical procedures constitute the mainstay of treatment. von Hippel-Lindau syndrome is a disorder of autosomal dominant inheritance that occurs in 1 in 40,000 births. The mean age at onset is in the fourth decade of life. The syndrome is inherited as a result of a germline mutation in a single allele of the VHL gene tumor suppressor gene located on chromosomal band 3p25–26.15,45,46 Sporadic loss of the remaining wild-type VHL allele provides the “second hit” necessary for tumorigenesis, most commonly via chromosome 3p deletion. Multifocal tumorigenesis is observed in multiple organ systems, with each tumor harboring an independent second VHL mutation. Renal manifestations include cysts and clear cell RCC tumors. Both tend to be multifocal and bilateral, and are found in the majority of patients with von Hippel-Lindau disease.15,45,46 Hundreds of independent clear cell cancers may be present in a single kidney, including dozens of macroscopic tumors. VHL syndrome patients are at high risk for chronic renal insufficiency because of the lifelong risk of multifocal RCC tumor development and need for repeat renal surgeries. As a result, VHL patients should undergo active surveillance until the largest tumor reaches 3 cm, at which time attempts may be made to resect all tumors in that kidney. Resection by enucleation without clamping of the main renal artery is recommended to maximize nephron sparing. Surgical candidates, particularly those with numerous tumors, are counseled as to the high possibility of local recurrence from de novo tumor formation and future ipsilateral surgery. The discovery of the VHL gene in hereditary clear cell RCC enabled the subsequent identification of a VHL mutation in sporadic clear cell RCC tumors (see above).47 Birt-Hogg-Dubé syndrome is a disorder of autosomal dominant inheritance. Signs and symptoms usually manifest in the fifth decade of life and include renal tumors and cysts, benign skin tumors (fibrofolliculomas) and pulmonary cysts, which can lead to spontaneous pneumothorax. The renal neoplasms may be multifocal and bilateral tumors and most often have pure chromophobe histology or a “hybrid” mixture of chromophobe and oncocytoma; infrequently, pure oncocytoma tumors may be present. Patients can present with several different tumor types within the same kidney and the presence of benign (oncocytoma) and malignant tumors within the same kidney should immediately prompt the suspicion of Birt-Hogg-Dubé (BHD) syndrome. The BHD gene mutated in this syndrome encodes the protein Folliculin and is located on chromosome 17p11.2.15,45,46,48,49 The BHD gene appears to have the characteristics of a loss-of-function tumor suppressor gene.50 Folliculin has unknown function but is found in complexes with adenosine monophosphate-activated protein kinase, the major sensor of cell energy and a negative regulator of the mammalian target of rapamycin (mTOR) pathway. Recently, multiple studies have implicated Folliculin in adherens junction formation and signaling.51,52 Tuberous sclerosis is a syndrome of autosomal dominant inheritance, with two genes identified, TSC1, located on 9q34, and TSC2, located on 16p13.3. It affects 1 in 6000 people and is usually diagnosed at birth.15,45,46 This syndrome encompasses multiple organ systems, including dermatologic, cardiac, pulmonary, and renal. Skins lesions include facial angiofibromas, periungual fibroma, shagreen patches, and hypopigmented macules. Patients also develop cardiac rhabdomyomas, pulmonary lymphangioleiomyomatosis, retinal hamartomas, subependymal nodules, and giant cell astrocytomas. The renal manifestations include bilateral and multifocal angiomyolipomas (AMLs) and less commonly clear cell renal carcinoma. In contrast with spontaneous AML, AML in this setting can be associated with a low risk of occult RCC (1%). The TSC1 and TSC2 gene products inhibit activation of mTOR signaling, a major promoter of protein synthesis and cell growth. Hereditary papillary renal cell carcinoma (HPRCC), inherited as an autosomal dominant trait, is caused by mutations in Met protooncogene on chromosomal band 7q31–34.15,46,53 It is characterized primarily by bilateral, multifocal papillary type I RCC. These tumors are not aggressive and rarely metastasize. Age at onset is around the fifth decade. The Met oncogene encodes a membrane tyrosine kinase that, in HPRCC, harbors an activating mutation in the kinase domain. Hyperactivation of the Met oncoprotein leads to upregulation of several intracellular signaling pathways involving Akt, Rac, and MAP kinase. Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is a disease of autosomal dominant inheritance. The gene for this disorder localizes to chromosomal band 1q42.3–43 and has been identified as fumarate hydratase (FH).15,45,46 Age at onset is between the third and fourth decades of life. The syndrome consists of cutaneous leiomyomas, uterine leiomyomas (fibroids), and papillary type II RCC tumors with high metastatic potential, even when small in size. Unlike the VHL patients, in whom delay in surgical treatment is usually the rule until the largest tumor reaches 3 cm, there should be no delay in treatment of solid renal lesions in these patients. Cystic lesions in these patients should be followed with close surveillance and early surgical intervention for any radiographic development of a potentially solid component. Thirty percent of patients have solitary and unilateral renal tumors.45,46 The FH gene functions as a tumor suppressor, with loss of the second allele detected in kidney tumors. The wild-type gene encodes an enzyme in the Krebs cycle catalyzing fumarate conversion into malate. Loss of the FH enzyme leads to accumulation of fumarate, which has been suggested to promote carcinogenesis through indirect stabilization of transcription factors HIFα and NRF2.39,40,54 Distinct from HPRCC and HLRCC, Malchoff and colleagues have described a three-generation family with five cases of papillary thyroid carcinoma and two cases with papillary renal neoplasia.53 With the use of linkage analysis, these investigators demonstrated that the fPTC/PRN phenotype was linked to 1q21.53 They characterized a distinct inherited tumor syndrome that may establish a link between papillary RCC and familial papillary thyroid carcinoma.53 Hereditary renal cell cancer associated with melanoma has been recently described. The TFE3 gene mutated in sporadic RCC (see “Diagnosis of Renal Cell Carcinoma”) is one of four members of the MiT family of transcription factors; although TFE3 mutations have not been identified in hereditary RCC syndromes, the related MiT member, MiTF, was shown to have a specific amino acid substitution associated with hereditary RCC tumors associated with melanoma.55 This substitution confers hyperactivation of MiTF transcriptional activity by preventing its sumoylation and degradation. Histologic features of these MiTF renal cancers are yet to be characterized. As the use of imaging methods has become more widespread, the frequency of incidental detection of RCC has increased. Patients with RCC typically present with a mass involving the kidney that is suggestive of the diagnosis. Nephrectomy is the most effective therapy for RCC that is confined to the kidney and should be used both diagnostically and therapeutically in most patients who are suitable surgical candidates. In certain clinical settings, percutaneous biopsy of a renal mass should be considered. In a retrospective study of 115 consecutive percutaneous biopsies performed on renal masses in 113 patients, investigators found percutaneous biopsy to be of high sensitivity in three clinical groups: patients with a known malignancy (N = 55), patients with no known malignancy and suspected unresectable tumor (N = 36), and nonsurgical patients with a mass suspected to be a resectable RCC (N = 8).57 Percutaneous biopsy of renal masses appears to be safe, carrying only a minimal risk of tumor spread. Urologists should consider increasing the indications for renal biopsy of small renal masses that appear to be RCC, especially in elderly and surgically unfit patients. The standardization of a sheathed biopsy technique by interventional radiology has alleviated fears of tumor seeding through the biopsy tract. With more experience and follow-up preoperative biopsy, this strategy has the potential to decrease unnecessary treatment, because up to a third of small renal masses are now reported to be benign at surgery. Percutaneous biopsy also may allow better selection of renal tumors for active surveillance and minimally invasive ablative therapies. However, there are certain histologic subtypes that cannot be easily distinguished by percutaneous biopsy. Oncocytoma for instance can only be diagnosed by resection, as rarely clear cell carcinoma may harbor regions of oncocytic cells, which are indistinguishable from oncocytoma with a single-needle core. In cases where oncocytoma may be suspected (e.g., in a patient with prior multifocal oncocytomas in the contralateral kidney), several staged biopsies of the mass can be performed to increase the confidence in the diagnosis. A RCC is unlikely to have three separate biopsies all positive for oncocytic cells only without any clear cell components. Finally, initial therapy for mRCC may potentially be stratified by histologic subtype and, in the future, molecular characteristics. The tumor–node–metastasis (TNM) system is a dynamic staging method that continually changes on the basis of new evidence from clinical studies (Table 82-2).58 This staging system is a method of stratifying patients with cancer and is based on data from large multicenter studies with large numbers of patients and a good level of evidence. Despite continual revisions to the methodology to incorporate new clinical evidence, however, the optimal RCC patient stratification using the TNM staging system remains controversial, and further revisions probably will be needed. Revision of the TNM staging system for RCC is likely to result in the simultaneous update of the integrated prognostic systems currently used with this traditional method of staging. Table 82-2 Tumor–Node–Metastasis (TNM) Staging System for Renal Cell Carcinoma
Cancer of the Kidney
Incidence and Risk Factors for Sporadic Renal Cell Adenocarcinoma
Pathology
Sporadic RCC 2004 WHO Classification
Histologic Tumor Type
Prevalence (%)
Cytogenetic Findings
Clear cell RCC
70
3p25-26 (VHL)
Papillary RCC
10-15
Trisomy of chromosomes 7 and 17, loss of Y chromosome, 7q34 (c-Met)
Chromophobe RCC
4-6
Loss of multiple chromosomes: 1, 2, 6, 10, 13, 17, 21
Multilocular cystic RCC
<1
Extracellular matrix gene
Collecting duct carcinoma
<1
Loss of multiple chromosomes: 1, 6, 8, 13, 14
Medullary carcinoma
<1
Sickle cell trait
Mucinous tubular and spindle cell carcinoma
<1
Neuroblastoma-associated RCC
<1
Xp11.2 translocation–TFE3 carcinoma
1-2
Translocations involving Xp11.2 (TFE3)
Unclassified lesions
4-5
Hereditary RCC: Syndromes and Histologic Tumor Types
Hereditary Syndrome
Chromosome (Gene) Abnormality
Histologic Type of Renal Tumor
Systemic Manifestations
von Hippel-Lindau
3p26 (VHL)
Clear cell RCC
Retinal angiomas, central nervous system hemangioblastomas, pheochromocytoma
Hereditary papillary RCC
7q34 (Met)
Type 1 papillary RCC
None
Hereditary leiomyoma RCC
1q42–43 (FH)
Type 2 papillary RCC
Cutaneous and uterine leiomyomas
Birt-Hogg-Dube syndrome
17p11.2 (BHD)
Chromophobe RCC, oncocytoma, hybrid tumors
Skin lesions, lung cysts
Genetics and Epigenetics of Renal Cell Carcinoma
Sporadic Renal Cell Carcinoma
Clear Cell Renal Cell Carcinoma
Papillary Types I and II Renal Cell Carcinoma
Chromophobe Renal Cell Carcinoma
TFE3-Fusion Renal Cell Carcinoma
Familial Renal Cell Carcinoma
Diagnosis of Renal Cell Carcinoma
Staging Systems for Renal Cell Carcinoma
Staging
Classification
1987
1997
2002
Tumor
T1
Tumor ≤2.5 cm, limited to kidney
Tumor ≤7 cm, limited to kidney
NA
T1a
NA
NA
Tumor ≤4 cm, limited to kidney
T1b
NA
NA
Tumor >4 cm and ≤7 cm, limited to kidney
T2
Tumor >2.5 cm, limited to kidney
Tumor >7 cm, limited to kidney
Tumor >7 cm, limited to kidney
T3
Tumor extends into major veins or invades adrenal or perinephric tissues, but not beyond Gerota fascia
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access