Epidemiology
It is estimated that 15,780 new cases of cancer and 1,960 cancer-related deaths occurred among children and adolescents below age 20 in the United States in 2014.
1 Tumors of the head and neck region account for 5% to 10% of all childhood malignancies.
2,3 Similar to other childhood cancers, the incidence of head and neck malignancies in the pediatric age group has demonstrated apparent increases in the United States in recent decades.
2 Given the potential for pediatric head and neck tumors or their treatment to adversely affect critical structures, functional outcomes, and quality of life, coupled with the rarity and diversity of many such tumors, multidisciplinary team management with expertise specializing in pediatric cancer is obligatory.
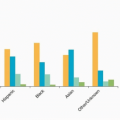
In an analysis using the National Cancer Institute’s population-based tumor registry, Surveillance, Epidemiology, and End Results (SEER), 3,050 children younger than age 19 years with head and neck tumors were identified from 1973 through 1996. Dominant histologic categorizations included lymphoma (27%), neural tumors (including neuroblastoma and retinoblastoma, 23%), thyroid cancers (21%), and soft tissue sarcomas (12%). The most common individual diagnoses were papillary thyroid carcinoma (
n = 537), Hodgkin lymphoma (HL) (
n = 515), retinoblastoma (
n = 497), non-Hodgkin lymphoma (NHL) (
n = 301), and rhabdomyosarcoma (
n = 239). Unlike cancers of the head and neck among adults, squamous cell carcinomas in pediatric patients are rare, accounting for fewer than 2% of cases.
2 Another recent analysis, looking only at sarcomas of the head and neck in the SEER database from 1973 through to 2010, identified 1,244 pediatric cases (including up to age 19 years) and 11,481 adult cases, with 10-year survival of 71% in pediatric patients compared to 61% in adult patients.
4 In this analysis of head and neck sarcomas, the most commonly reported histologies were rhabdomyosarcoma (48%), malignant fibrous histiocytoma (MFH, 11%), osteosarcoma (8%), and Ewing sarcoma (6%).
4The age of presentation can provide important initial guidance for conditions likely to be malignant arising in the head and neck. From the SEER database, retinoblastoma, neuroblastoma, germ cell tumors, and rhabdomyosarcoma are among the most common conditions among neonates and infants.
2 Among children below age 10 years, one can see various conditions, including NHL and HL. Among children and adolescents above age 10 years, dominant malignancies of the head and neck include thyroid cancer, Hodgkin and non-Hodgkin lymphoma, and melanoma.
2 Certain tumors are rarely seen outside of their typical age of presentation, including malignant germ cell tumors involving the head and neck, which are mainly seen only in neonates or infants,
5 and retinoblastomas, which are rarely seen in children older than 5 years.
Of note, clinical reviews of malignancies of the head and neck do not consistently include important differential diagnoses such as retinoblastoma in the pediatric age group, and leukemia presenting with chloromas. In addition to primary cancers arising in the head and neck, conditions such as malignant germ cell tumor, lymphoma, and neuroblastoma may also metastasize to the nodes, soft tissue, or bones of the head and neck.
Secondary malignant tumors of the head and neck, that is, those resulting from prior therapy including radiation therapy and alkylating chemotherapy agents, account for about 0.8% of tumors of the head and neck in the pediatric age group
2 and may include thyroid carcinoma, osteosarcoma, MFH, and salivary gland tumors.
6,7
Diagnostic Evaluation: An Overview
As most masses in the head and neck in pediatric age group are benign, careful clinical history and physical examination with additional workup when appropriate are warranted to diagnose patients with malignant lesions. Conditions to be considered include lesions primarily arising in the head and neck, those that are metastatic to the head and neck, and secondary malignancies. Primary head and neck lesions can include congenital or developmental lesions, inflammatory or infectious masses, benign tumors, or malignant tumors
(Table 24.1).
Assessment of a child with a mass in the head and neck should start with a detailed clinical, past medical, and family history, immunization history, and recent and current medications. History should detail the onset, nature, and timing of any changes associated with the mass, along with potential exposures and risk factors for infection (e.g., tuberculosis), travel, and exposure to animals (e.g., zoonoses).
Congenital or developmental lesions may first be evident in the early postnatal period, but in some cases they can manifest later in life, with gradual growth over time or with concomitant secondary infection. Benign tumors can similarly sometimes be brought to medical attention in the setting of concurrent infection. Whereas inflammatory/infectious etiologies are typically first considered for masses that acutely enlarge and are associated with acute fever, tenderness, or skin changes such
as erythema or warmth, red flags should prompt consideration of an underlying malignancy. Potential red flags include firm, fixed/immobile or large (>1.5 to 2 cm) masses; lymph nodes in atypical locations (e.g., supraclavicular) or multiple locations; masses initially queried to be inflammatory or infectious in nature that do not respond to initial empiric management or persist beyond 4 to 6 weeks; or constitutional symptoms such as malaise, weight loss, anorexia, and unexplained fevers or night sweats.
Relevant clinical and functional impairments to assess on history and to observe on examination may include abnormal eye movements; distorted vision; proptosis; Horner syndrome; cranial nerve palsies; paresthesias; recurrent epistaxis; increasing nasal congestion; evidence of recent suspected/confirmed infections; loss of smell; stridor; dysphagia and odynophagia; impaired gag; trismus; deviated tongue; dental health; change in tooth position; change in voice or hoarseness; otorrhea; progressive tinnitus or hearing loss; otalgia; vertigo; neck tilt or torticollis; facial and/or neck swelling; deviated trachea; increased vascular markings, pulsatility, or overlying skin changes; or features of hypothyroidism or hyperthyroidism.
Physical examination should also include evaluation for movement of the mass with tongue protrusion (e.g., thyroglossal duct cysts, which typically move upward with tongue protrusion) and include a full systemic examination for systemic lymphadenopathy, with particular vigilance for lymph nodes in atypical sites such as the supraclavicular area or that are firm, fixed, and significantly enlarged; ophthalmologic examination; assessment of tonsillar tissue; oral mucosal examination; thyroid examination; cardiorespiratory examination; abdominal examination for masses or hepatosplenomegaly; genitourinary examination; skin examination for evidence of petechiae or purpura or neurocutaneous markings; dysmorphic features; detailed neurologic examination; and note of any current or impending functional impairment from mass effect.
Any concern for potential airway compromise must be immediately evaluated and managed as an oncologic emergency. As tumors in the head and neck may expand into cavities such as the maxillary sinus with relatively limited innervation, pain is not necessarily a dominant symptom and more likely becomes prominent with nerve compression or periosteal impingement.
8 Potential intracranial extension, with intraparenchymal brain metastasis or leptomeningeal disease, can also be seen in children and may also be associated with varied acute neurologic symptoms, warranting careful evaluation and management.
8,9 Careful clinical and radiologic assessment as appropriate for a potential mediastinal mass should also be pursued, and managed as a medical emergency, given the risk of acute cardiorespiratory decompensation with sedation or suboptimal body positioning. Once oncologic emergencies have been ruled out or definitively managed, a broad differential diagnosis should be considered.
Targeted laboratory studies should be considered to further evaluate suspected conditions based on initial history and physical examination. Evaluations for persistent adenopathy not responsive to empiric antibiotics may include considerations for Bartonella henselae (catscratch), Epstein-Barr virus (EBV), cytomegalovirus, toxoplasmosis, histoplasmosis, tuberculosis, and human immunodeficiency virus as appropriate.
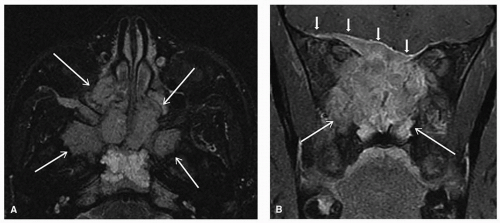
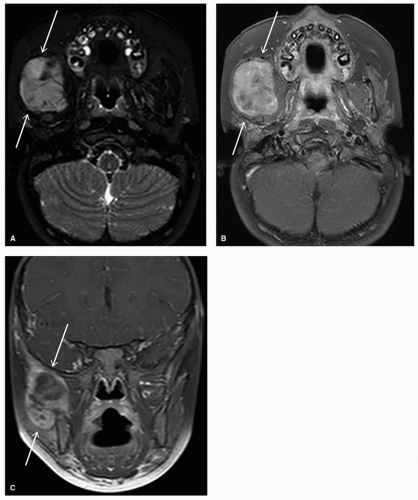
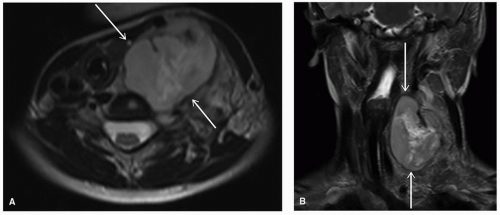
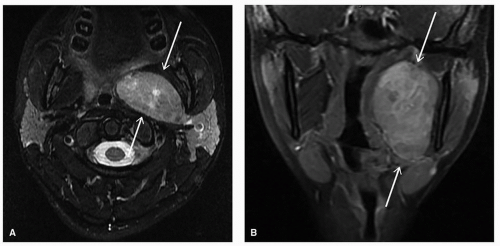
Targeted imaging should also be considered to help with diagnosis and further management planning, with sample modalities outlined in
Table 24.2. For children (up to age 14)
with a mass in the neck or adenopathy, the American College of Radiology (ACR) Appropriateness Criteria recommends the use of neck ultrasound as the most appropriate modality, followed by computed tomography (CT) of the neck (with contrast) and magnetic resonance imaging (MRI) of the neck (without and with contrast).
10 Although intravenous contrast is typically recommended with cross-sectional imaging to help characterize the margins of the lesions and abnormally enhancing lesions that may not be pathologically enlarged, this should be considered in conjunction with a specialist team that includes a radiologist. For instance, noncontrast CT is recommended in the setting of suspected salivary gland enlargement due to a sialolith,
10 whereas for a suspected mass in the thyroid, use of iodinated contrast agents is typically avoided or used judiciously only after discussion as a specialist team, as iodine uptake in the thyroid may affect timing of subsequent diagnostic or therapeutic steps.
A chest radiograph, which is readily available, can be useful for particular circumstances. These can include situations when one suspects a mediastinal mass, an oncologic emergency that requires care in patient positioning during the evaluation, as well as for suspected lesions extending into the chest or for gross metastatic disease.
Specialized imaging such as
18F-fluorodeoxyglucose positron emission tomography (FDG-PET), CT or magnetic resonance angiography, or other body imaging is typically reserved for confirmed malignancies in the setting of staging or follow-up evaluation. Key considerations for imaging in children with head and neck malignancies are outlined in
Table 24.3, with more detailed depictions in the sections and references below.
11,12,13,14,15,16,17 In all cases, the concept to “image gently” and expose children to “as low as reasonably achievable” (ALARA) levels of ionizing radiation in imaging should guide practices.
16,18Early consultation with a multidisciplinary specialist team, including providers in pediatric surgery and pediatric hematology/oncology, is warranted for any concerns of potential malignancy.
Fine-needle aspiration (FNA) may be considered for initial tissue evaluation of selected neck lesions of children, with sensitivity ˜90% or higher, and specificity ˜85%, where the most likely diagnosis is reactive lymphadenopathy.
19,20 Advantages include its minimally invasive nature compared to an open biopsy, relatively low cost, and potential avoidance of general anesthesia in some children.
3,19,20 Anticipatory multidisciplinary planning prior to any potential biopsy is recommended, including at least the surgical, radiology, and pathology teams, as image guidance and rapid on-site processing could critically impact diagnostic yield. With the aid of experienced pediatric cytopathology team members, FNA or core needle/open biopsy specimens can be processed for rapid interpretation with direct smears and cell block preparation for routine hematoxylin and eosin-stained slides, as well as ancillary studies, which can include cultures, special stains for acid-fast bacilli or fungi, flow cytometry, or fluorescent in situ hybridization.
20 Core needle or open biopsies are recommended when malignant disease is likely, bony lesions are suspected, or preserved tissue architecture or more complete evaluation of suspected heterogeneous tissue (which may be missed by selective FNA sampling) are required for diagnostic confirmation or where FNA sampling was insufficient, indeterminate, or inconsistent with the clinical impression.
8,20
Clinical Management: An Overview
Optimal management of malignant tumors in the head and neck conditions requires a coordinated multidisciplinary team with familiarity and dedicated expertise in working with children with these rare and complex cancers. Inherent in the very location of tumors of the head and neck and their proximity to multiple vital structures, this group of tumors warrants anticipatory evaluation and management of potential airway compromise and special anesthesia needs and potential functional implications of the mass in its natural course, as well as implications secondary to diagnostic and therapeutic procedures, while considering the child’s developmental growth and function.
Functional sequelae to be considered and optimally prevented may include more immediate concerns involving compromised swallowing and the risk of aspiration or ophthalmologic complications,
21 as well as longer-term implications of the cancer or therapy on orofacial development and dentition,
22,23 jaw function,
24 cosmesis, and cognitive function. For instance, patients with a history of dysphagia would benefit from clinical bedside swallowing evaluation by speech language pathologist, as well as barium swallow examination if clinically indicated.
Vigilant supportive care is also required to evaluate and address the known toxicities of cancer therapy that may be exacerbated by the location of the tumor of the head and neck, including oral mucositis. Young patients require nutritional and growth monitoring and may benefit from feeding tubes for enteral nutrition as well as for selected administration of medication. As many effects of the cancer therapy may initially become manifest or progress months or years beyond the treatment period, long-term follow-up for potential late effects is warranted.
Updated general guidance for the core personnel and resources essential to provide care for children and adolescents with cancer has been recently articulated by the American Academy of Pediatrics. Outcomes for children with malignancy have been demonstrated to be better when core management is at least initiated by specialist pediatric cancer centers.
Typical specialist multidisciplinary team members engaged closely with families of children with malignant masses in the head and neck should include pediatric physicians, nurses, pharmacists, social workers, and other allied health team members working in hematology/oncology; surgery, otolaryngology, and neurosurgery; ophthalmology; dental oncology and orthodontics; radiation oncology; plastic and reconstructive surgery; radiology and diagnostic imaging; pathology; nutrition; speech/language pathology; and child life. In many cases, the expert consultative input of other pediatric team members in anesthesiology, critical care, endocrinology, genetics, infectious diseases, neurology, ocular prosthesis, palliative care, psychology, rehabilitation specialists including occupational and physical therapy, and team members experienced in late effects of therapy is also invaluable. In all cases, the pediatric patient, the family, and the community-based or primary care team members engaged by the family remain central partners with the multidisciplinary specialist team in decision-making and family-centered care delivery.
Surgical Considerations: An Overview
Surgical management of pediatric age group cancer of the head and neck has undergone many significant changes during the past two decades. Most of these changes have occurred as a result of technologic advances, including better imaging, more precise delivery of radiation, advances in surgical technique, and a better understanding of anatomy. The improved understanding of anatomy has had its greatest impact on the development of better and more reliable reconstructive techniques that allow us to perform surgery that, in the past, would have been impossible. We can now be more certain that the defects that we create surgically can, for the most part, be reliably reconstructed. However, in the pediatric age group, cancer of the head and neck is uniquely affected by the need to account for the growing and developing facial skeleton and associated structures. This has a particular impact in children’s dentition, which owing to its continued growth until skeletal maturity often requires a staged approach to reconstruction.
Evaluation for surgery is first dependent on accurate tissue diagnosis, requiring incisional or excisional biopsies for the vast majority of solid tumors. Exceptions include salivary or thyroid masses, for which fine needle aspiration is an option. Subsequent staging evaluation includes routine laboratory studies (complete
blood count, basic metabolic panel, liver function tests, lactate dehydrogenase [LDH]), CT scan of the chest, and in many cases positron emission tomography (PET) scan. Unlike in cancer in adults, pediatric resections must balance the extent of surgical resection with functional and aesthetic concerns, and thus, radical resection with negative margins (R0) resections is not always possible. Some pediatric tumors, in fact, are so responsive to nonsurgical therapies or are so indolent in nature (e.g., desmoid tumors) as to make resections unnecessary. As with adult malignancies, patients are frequently candidates for either chemotherapy and/or radiation therapy in either a neoadjuvant or an adjuvant setting. Depending on the pathology of the disease, timing of such treatment may vary. Finally, reconstructive concerns share many similarities with cancers in adults, but have specific differences with regard to the developing pediatric facial skeleton and associated structures.
Role of Radiation Therapy: An Overview
Radiotherapy is a critical component of the multimodality management in the pediatric age group of cancer of the head and neck. Although some tumors involving the head and neck are surgically accessible, complete extirpation of lesions occupying anterior craniofacial spaces or the base of the skull is often impossible without significant morbidity necessitating a management plan that relies on nonsurgical modalities either as surgical adjuncts or for definitive tumor control. Microscopic residual disease is a source of local failure that can be addressed via the use of adjuvant radiotherapy or chemoradiotherapy. Clinical studies of pediatric head and neck cancer seek optimal combinations of multimodality therapy in order to reduce late effects while simultaneously increasing rates of long-term disease control.
Conformal radiotherapy with or without intensity-modulated radiation therapy has been the primary mechanism of radiation dose delivery during the past two decades.
25,26 Advances in software now routinely employ computation-heavy, cost-benefit function analyses of beam modulation to address irregular structures within the body, particularly when they are concentrically contained within or encapsulated by normal organs with dose-limiting sensitivity to radiation effect.
27,28 Furthermore, physiologic motion is minimal within the head and neck region, allowing greater confidence in patient setup and targeting the tumor bed. Although some advances, like respiratory gating and breathing management (deep inspiration breath hold), have been developed to address organ and target volume motion, most treatment of the head and neck in the pediatric age group can be accomplished without resorting to these specialized measures.
29 Prosthetics in the form of intraoral molds and the use of thermoplastic head and neck immobilization devices frequently serve as positioning aids, both internally and externally for the pediatric oropharynx and head/neck, respectively.
30,31 Fiducial markers can also be implanted to further aid in targeting when extraordinarily high precision is required to spare critical structures skirting radiation dose thresholds.
Frequently, these tumors afflict young children and infants requiring sedation to achieve adequate immobilization suitable for treatment. Most institutions choose to sedate with propofol and avoid airway management when possible, although occasional management of the airway is required to ensure patient safety through a full course of radiation, as determined by the anesthesiologist.
32Proton therapy has recently become central to the discussion of pediatric radiotherapy due to the promise of reduced toxicity; larger-scale clinical trials designed to demonstrate the potential advantages are ongoing. Although a major objective of proton therapy is reduction of toxicity, protons do not inherently promise a more efficacious means of disease control (unless normal tissue sparing through improved tumor targeting is enhanced such that dose escalation of residual tumor can result in improved disease control).
33 At this time, precision targeting with proton beam is still developing as a routine component of clinical practice due to various factors, including cost and accessibility. Proton therapy requires prospective study design to provide evidence for the appropriate indications and degree of benefit that may be obtainable.