1
Introduction
Epidemiological evidence supports an association between diabetes and cancer and raises a number of questions related to this issue such as (i) Is this a positive or negative association? (ii) is diabetes associated to all kinds of cancers and with similar magnitude? (iii) all types of diabetes are associated to those cancers? (iv) does the association rely on treatments or on the disease? (v) are there antidiabetic treatments that promote or prevent cancers? (vi) do antineoplastic treatments promote diabetes? (vii) what appears first diabetes or cancer? (viii) how long does it take between diagnoses of diabetes and that of cancer or vice versa? (ix) how does diabetes alters morbidity and mortality in cancers? (x) how does cancer alter diabetes? (xi) how does diabetes alter cancer screening? and (xii) how can knowledge on this issue improve the quality of life of diabetic patients?
Concerning the first two questions (i–ii), diabetes is generally associated to cancer, but the term cancer groups a variety of very different diseases, which obliges to make separate analysis for distinct cancers. Not all cancers are associated to diabetes, some are unrelated. Some cancers are more frequent in the diabetic population (direct association) and perhaps are favored by diabetes conditions, and other cancers are less frequent among diabetic patients (inversely associated) and may be interfered by diabetic conditions. This suggests the existence of “discrete factors” in diabetes with differential influence on the appearance of certain cancers either with protective or enhancing effects. Thus, diabetes and cancer are multifactorial diseases that share metabolic, hormonal, immune, and microbioma abnormalities, and epidemiological studies establish clear associations between diabetes and different site-specific cancers.
Regarding question (iii), that is: all types of diabetes are associated to those cancers? The type of diabetes marks the association to cancers. In general, type 1 diabetes (T1D) is associated with less site-specific cancers than type 2 diabetes (T2D). However, since less than 5% of diabetic patients suffer T1D, it is more difficult to reach statistical significance, and despite that some cancers associate with T1D and not with T2D. Thus, differences between both diabetes types must be important for cancer initiation.
The variability in the association of site-specific cancers with T1D and T2D suggests that more than one factor from diabetes contributes and interact with local factors (additive or oppositely, depending on the site) to favor or interfere with initiation or progression of a given cancer. This would explain differential associations of diabetes with stomach, colorectal, or prostate cancers.
This leads to the next three related questions: (iv) Does the association rely on treatments or on the disease? There is no clear answer to this question yet; diabetes may contribute with metabolic, hormonal, immune, and microbioma abnormalities, many of which are shared with cancers. Moreover, antidiabetic treatments may interfere with cancer biology and antineoplastic treatments, which lead to the next questions: (v) Are there antidiabetic treatments that promote or prevent cancers? Certain insulin preparations have been related to increased frequency of pancreatic, liver, lung, and prostate cancers and will interfere with antineoplastic treatments targeting tyrosine kinases, whereas the biguanide metformin that lowers blood glucose and its cellular respiration has been inversely associated to other cancers. Likewise, the answer to question (vi), that is, do antineoplastic treatments promote diabetes?, is yes. Some cancer treatments such as glucocorticoids, tyrosine kinase inhibitors, or checkpoint inhibitors induce metabolic derangements such as high-grade hyperglycemia leading to diabetes. Answering what appears first, diabetes or cancer may give a clue as to what factors link these diseases (vii). However, diabetes-cancer relationships are bidirectional with diabetes being diagnosed first in most cases and later appearance of cancer. Less frequently a cancer appears, and diabetes develops subsequently as a consequence of cancer evolution or of antineoplastic treatments.
Regarding the time lapse (viii) between the diagnosis of diabetes and that of an associated cancer or vice versa, there are still few studies that consider this point and the data is not conclusive, perhaps because both diabetes and cancer may be diagnosed after a period of silent disease. What appears clear is that diabetes alters morbidity and mortality in cancers (ix) and co-occurrence of both diseases worsens the prognosis and increases the morbidity and mortality even in cancers that appear inversely associated to diabetes. Of course, cancer will also alter diabetes (x) since high levels of circulating nutrients including glucose or lipids will be consumed by cancer cells, and cancer progression may induce peripheric insulin resistance and increase the levels of circulating IGF-1. It appears logical that if certain cancers are more frequent in the T2D population, the presence of diabetes should increase/intensify cancer screening (xi). However, at present, there are no specific guidelines that promote a closer screening in diabetic patients for cancers positively associated such as colorectal or endometrial even though their incidence is increasing. Knowledge on these issues should improve the quality of life of the vulnerable diabetic population (xii) extracting good from evil. Selective directed screening should improve early detection of cancers associated to diabetes, and with it its early removal ensuring successful treatment.
Identification of the individual factors that link diabetes to cancer should be a priority for prevention and management. In this chapter, we review the current data to cover the state of the art pertaining this interesting issue and revise the potential factors that contribute to the diabetes-cancer association.
2
Epidemiological association between diabetes and cancer
The epidemiological association between diabetes and cancer is concerning because diabetes incidence is increasing, threatening with increases in the incidence of specific cancers. According to the World Health Organization (WHO), the incidence of diabetes doubled between 1980 and 2015 and continues increasing [ , ]. In depth studies will help to predict increases in specific cancers, help to develop strategies for prevention, and guide the search and identification of contributing factors. Strategies to prevent diabetes acquire maximal importance, since coexistence of diabetes and cancer worsens the prognosis even in cancers inversely associated.
The main risk factor to suffer T2D is obesity, but the incidence of obesity quadruplicates diabetes incidence, meaning that not all obese people get T2D but most T2D patients are obese. Similarly, not all diabetic patients get cancers, but the frequency of some site specific cancers is hauntingly superior in the diabetic population and the number of cancer cases is predicted to increase by 70% between 2015 and 2030 [ ]. For example, colorectal cancer incidence is predicted to increase and is already the second/third more prevalent in women/men respectively and a major cause of cancer-related deaths [ , ]. Fig. 15.1 updates a systematic review [ ] selecting the best controlled meta-analysis to analyze the diabetes-cancer association data between 2006 and 2021 [ ]. The bar diagram draws the general perspective (bars to the right represent direct, and to the left, inverse associations) where the length is proportional to the strength of each association. Numbers on the right detail the relative risks with their 95% confidence interval. This figure puts into numbers the answer to the first three questions: the association diabetes-cancer can be positive or negative, the magnitude is different in distinct cancers and differs for T1D or T2D.
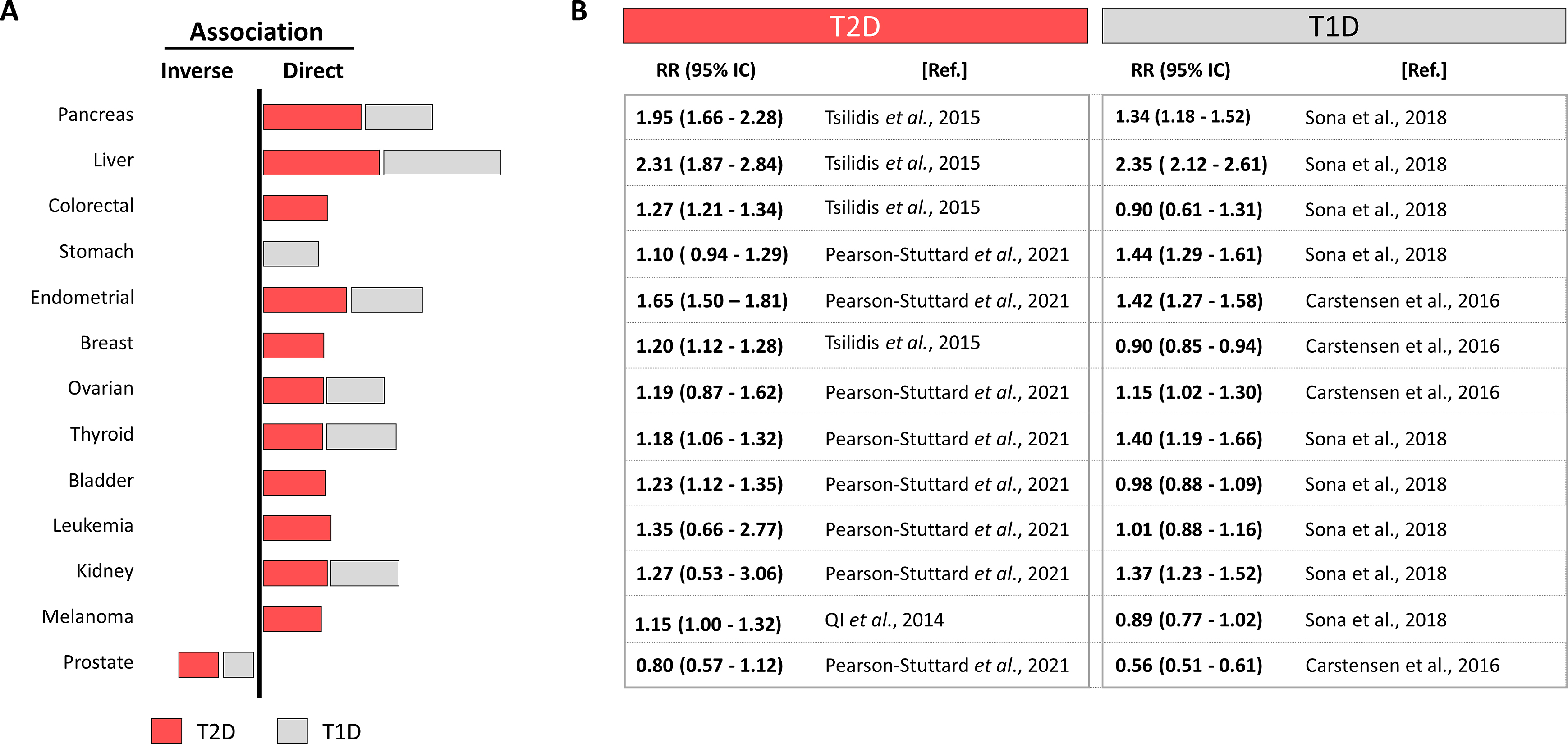
Interestingly, many cancers, as frequent as colorectal, breast, or melanoma, are directly associated with T2D but not with T1D, suggesting that contributing factors present in T2D are absent in T1D. This, together with the different strength of the associations with site-specific cancers suggests that individual contributors (positive or negative) from T1D or T2D interact differently in different sites. Notably many cancers associated to T2D are also directly associated to obesity, but not to T1D [ ]. Nevertheless, T1D represents only about 5% of all diabetes, imposing a difficulty to recruit patients to reach statistical power, which leads to more scarce studies. The same principle applies for cancers with low incidence, where finding patients with diabetes challenges the possibilities to reach statistical power and their association remains not established.
The next three questions posed in the introduction concern the nature of factors that may link diabetes to cancers. In addition to the reciprocal influence of factors inherent to diabetes or cancer (metabolic, hormonal, etc.) which are analyzed in the next epigraph, antineoplastic treatments may drive diabetes and antidiabetic treatments may interfere or cooperate with cancer development or progression.
The most widely used antidiabetic agents are exogenous insulin preparations and the biguanide metformin, which lowers blood glucose and its respiration. The diversity of treatments (including secretagogues, incretins, inhibitors of glucose absorption or metabolism, and a variety of combinations) and their evolution with the disease complicates the analysis of their influence on cancer initiation and not less important, strong economic interest are at play. Insulin is the paradigm treatment for T1D and in long-standing T2D. Exogenous insulin was positively associated to some cancers (pancreatic, liver, colorectal), but alternative treatments that increase circulating insulin, such as sulfonilureas, did not associate, perhaps the high levels of exogenous insulin act through related receptors (IGF-1) with preferential mitogenic signaling or the subcutaneous injection plays a role, see Ref. [ ] for review. Future studies will explain these differences. The main pitfall of studies that analyze the association of insulin treatments with cancers is the consideration of all cancers as one disease, since different cancers are different diseases and their frequency and their associations with T2D are uneven or opposite. Interestingly, treatment with metformin, widely used as first-line treatment for T2D, was inversely related to colorectal and other cancers by contrast with insulin and encouraging many (still running) clinical trials as adjuvant in cancer treatments. However, the results remain controversial, and it is unknown if the potential protective effect are exerted by controlling glucose homeostasis at the liver or by interfering with glucose metabolism in cancer or stromal cells.
Antineoplastic treatments, remarkably, may lead to diabetes development. Glucocorticoids are often used as adjuvant antineoplastic therapies because of their beneficial effects on edema, pain, and as immunomodulatory. Glucocorticoids may induce hyperglycemia depending on the doses and its continuous or discontinuous administration. In addition, PI3K, Akt (and other tyrosine kinase), and mTOR inhibitors, generally termed PAM inhibitors and widely used to treat cancers, are associated to development of severe hyperglycaemia (>250 mg/dl) and hyperlipidemia (with high total and LDL-cholesterol) [ ]. This is achieved through inhibition of the tyrosine kinase receptors for insulin and IGF-1, which signal through mTOR to control cellular glucose and lipid uptake. Hyperlipidemia is often counteracted with additional treatment with statins adding more complexity to the analysis of diabetes-cancer association. A third class of antineoplastic termed checkpoint inhibitors (including targeting the PD-1/PD-L1 axis) is directed to enhance immune destruction of the tumor but has been evidenced to drive T1D in genetically susceptible individuals, in both animal models and in humans [ ].
Few epidemiological studies approach the time-lapse between diabetes and cancer diagnoses, which concerns the next two questions posed at the introduction, which are important to guide cause-effect studies. In most cases, cancer is diagnosed years after the diagnosis of diabetes, and the time interval between both diagnoses varies for different cancers according to the scarce studies that analyze the question. Cancer risk increases during the first 2 years after the diagnosis of diabetes. For colorectal cancer, the risk continues increasing up to 8–10 years to decline afterward, whereas for endometrial cancer, the risk stabilizes from 2 up to 10 years [ , ]. For pancreatic cancer, less frequent but more strongly associated to diabetes, its risk increases up to 20 years after diabetes diagnosis. In general, the longer the period during which risk increases, the bigger the magnitude of the association. In some pancreatic cancers, diabetes appears after the cancer, sometimes as a snitch that helps earlier diagnosis of a silent cancer. However, the cases in which cancer is diagnosed first and diabetes later are less frequent than vice versa. For other cancers, there is not a time pattern for the association, or the risk does not change with diabetes duration. The difficulty in these studies is to establish for how long have evolved diabetes or cancer by the time of diagnosis since both may appear silently for variable periods.
Pertaining the next three questions posed in the introduction and dealing with how cancer and diabetes alter each other, epidemiological data evidence that coexistence of diabetes with any cancer, even those inversely associated, worsens the prognosis. There are few studies that quantify this influence by Kaplan–Meier analysis to compare the diabetic versus nondiabetic population on a given cancer and separating overall survival (OS) and disease-free survival (DFS). For colorectal cancer, a study found that OS in diabetic patients was reduced by 18%, leading to a 5 year reduction in survival [ , ]. In support of these data, the T2D population was more frequently diagnosed with more advanced colorectal cancers (stage 2), whereas the nondiabetic population was more frequently diagnosed stage 1. Comparison of 976 colonoscopies of diabetic and nondiabetic patients revealed that polyps and higher dysplasia were more frequent in the T2D population [ ], thus strengthening previous epidemiological evidence. This finding was unexpected because the T2D population should be more closely followed up and highlights the importance of selectively screening the T2D population for the most frequently associated cancers as a preventive strategy.
Thus, the epidemiological evidence supports the association between specific cancers and diabetes and worsening of the prognosis by coincidence. This highlights the need to understand not only treatment interferences, but more importantly the potential of individual factors from diabetes contributing to cancer initiation and/or progression, which is revised in the next epigraph.
3
Factors that link diabetes to cancers
Silent neoplastic lesions evolve for years to progress to cancer, accumulating mutations in oncogenes and/or tumor suppressors, selecting the most advantageous mutations to coordinate metabolism and environment. Molecular signatures defined through cancer evolution are useful for cancer classification and choice of therapy [ ]. A variety of intrinsic factors from diabetes may interfere and facilitate cancer initiation and/or progression, Fig. 15.2 . Cancer cell transformation may obey to metabolism changes in particular cells. Cellular responses to the diabetic environment include genetic and epigenome lesions that may drive chromosome instability or mutations [ ]. Tumors originate after an aberrant interplay between metabolism, signaling, and gene expression, where mutations may appear as the cause or the consequence of tumor evolution. Mutations that occur at key genes for cell cycle/proliferation, apoptosis evasion, metabolic adaptations, or signaling intermediates, are required to develop cancer cell hallmarks and will be positively selected. Remarkably, many of these mutations aberrantly activate receptor tyrosine kinases (RTK) signaling pathways, which are also involved in diabetes, for example, through the receptors for insulin or IGF-1. RTK-driven mitogen activated protein kinases (MAPK) signaling is well known as aberrantly activated in many cancers, but most signaling pathways activated in cancer, such as WNT, JAK/Stat, Hippo, and Notch pathways, are critical for metabolism control [ ]. Note that both, aberrant metabolic control or mutational activation, of these pathways will prevent their physiological control and integration with other environmental signals. Metabolic adaptations required for cancer cells to adapt to rapid proliferation and apoptosis evasion require increased nutrient uptake, which is favored by hyperglycemia and hyperlipidemia in the diabetic environment and may lead to metabolic transformation [ ]. Notably, increased nutrient uptake by cancer cells is often accompanied by decreased uptake with a certain degree of insulin resistance in other tissues in advanced cancers. Fig. 15.2 .
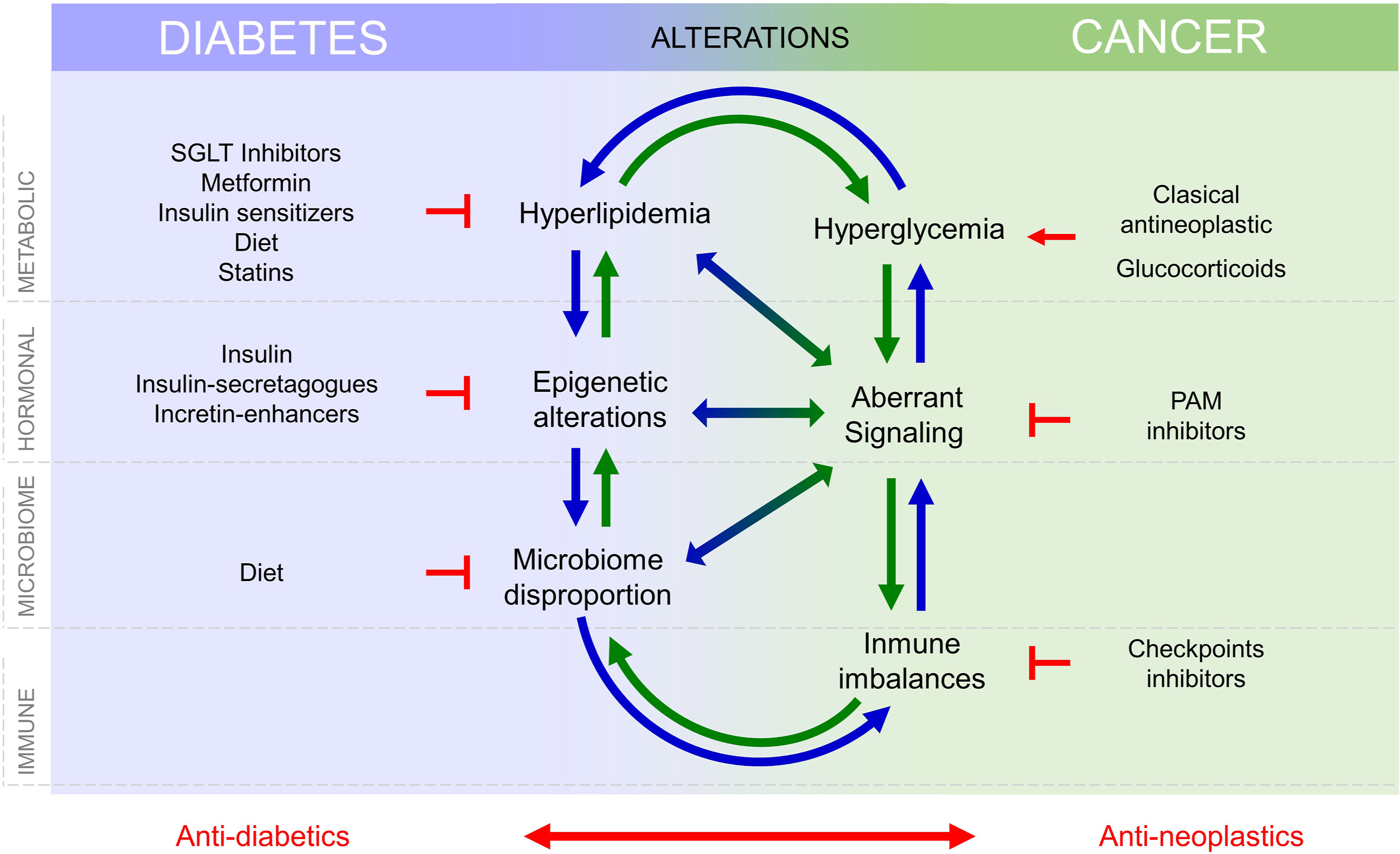
Intracellular nutrient levels and metabolites regulate and fine tune cytosolic signaling that control gene expression, as depicted in Fig. 15.2 , to regulate proliferation and survival. Conversely, aberrant gene expression programs activated in cancer control and reprogram cell metabolism [ ]. Thus, the interplay between metabolism, signaling, and gene expression, which is altered in diabetes may lead to cell transformation.
Diabetes is defined by metabolic and signaling aberrations. This includes excess circulating glucose and lipids, aberrant insulin, IGF-1 and IGF-2 signaling, and disproportion in adipokines such as leptin and adiponectin [ ]. These alterations lead to defects in vessel formation, and immune response, both integral parts of the tumor microenvironment (TME). Microbiome impoverishment, driven by the interplay between hyperglycaemia and immune defaults also influence cancer development [ ]. Altogether, increased circulating nutrient availability (glucose and lipids), aberrant growth factor signaling, altered angiogenesis, increased inflammation, and defects in immune surveillance as well as aberrant microbiome interactions may contribute independently or cooperatively for cancer initiation or progression, Fig. 15.2 . Their individual contribution must be identified and quantified to develop new therapies and adequate prevention strategies that lower the incidence rise of certain cancers that follows the increase in obesity-diabetes.
3.1
Metabolic reprogramming
Cancer cells exhibit increased nutrient uptake and will profit from excess circulating glucose and lipids in diabetes to overcome cell cycle checkpoints and apoptosis while also supporting morphological changes required for invasion and metastasis [ ].
Excess glucose provides fast energy and carbons for rapid proliferation, favoring metabolic transformation, which includes cytosolic and mitochondrial reprogramming. Enhanced glycolysis, pentose phosphate pathway (PPP) for NADPH regeneration plus reactive oxygen species (ROS) detoxification, and hexosamine pathway for glycosylation occur at the cytosol of cancer cells [ ]. Subsequent mitochondrial reprogramming reinforces anaplerotic pathways to supply the TCA intermediaries depleted by the deviation of carbons from glycolysis toward anabolic pathways [ ]. Mitochondrial reprogramming generates substrates that shuttle to the nucleus and alter the activity of epigenetic enzymes. Examples are citrate converted to acetyl-coenzyme A that provides acetyl groups for histones and transcription factors acetylation or alpha-ketoglutarate that alters DNA and histone methylation, epigenetic marks involved in defining gene expression patterns [ , ]. Mitochondrial reprogramming following metabolic adaptations causes fusion/fission imbalances and mitochondrial dysfunction and has been reported for T2D subjects (in muscle or pancreas) [ ] and in highly proliferative cancer cells [ ].
Excess circulating lipids, free fatty acids (FFA) and cholesterol are critical for membrane structure and function, energy storage, and signaling [ ]. Cancer cells exhibit increased lipid uptake and enhanced de novo lipid biosynthesis. Increased glucose uptake by cancer cells upregulates critical enzymes for lipid synthesis, which is critical for cancer cell proliferation and metastasis [ ]. Consistently, cholesterol storage in droplets is enhanced in cancer cells, providing energy for proliferation and migration. Thus, cancer cells will benefit from excess circulating lipids, characteristic of T2D.
Excess glucose and lipids modulate insulin, IGF-1, MAPK, Wnt, and other signaling pathways related to metabolic transformation. Glucose enhances Wnt and PI3K signaling, and in turn, these pathways increase glucose, lipids, and glutamine uptake firing a feed forwards cycle for metabolic reprogramming. Metabolism of excess glucose, lipids, and glutamine leads to accumulation of ROS that cause genetic lesions and substrates such as fumarate or alpha ketoglutarate available for epigenetic control [ ]. Abnormal epigenetic marks or patterns related to T2D include hyperacetylation patterns and hypo and hypermethylated genes related to metabolism and inflammation, which are very relevant for cancer [ ].
At the level of cancer cell phenotype, glucose excess enhances survival and proliferative phenotypes [ , ], and lipids enhance EMT for invasive and metastatic phenotypes [ ]; therefore, T2D excesses in glucose and lipids might contribute to phenotypic diversification of advanced tumors and with it to therapeutic resistances [ ].
3.2
Signaling alterations
Lack of insulin in T1D or insulin resistance in the more frequent T2D define the main signaling alteration in diabetes, although hormonal imbalances in the ratio leptin/adiponectin, in glucagon or even in cortisol may contribute. Hyperinsulinemia (from exogenous treatments in T1D or following insulin resistance in T2D) has been attributed a role in inducing cancer. Insulin is a growth factor that acts directly binding its receptor (IR), which is a tyrosine kinase (RTK) or indirectly through induction of insulin-like growth factors (IGF1 and IGF2) [ , ]. Insulin bound to its RTK signals through PI3K and MAPK for anabolism, survival, and mitogenic effects. IR comes in two flavors: IR-A (predominant in the embryo) and IR-B (in the adult). IR-A and IR-B signal slightly differently probably due to their specific distribution in different membrane domains. IR-A signals predominantly through MAPK for mitogenesis and IR-B through PI3K for anabolism and survival [ ]. Epithelial tumor cells increase their IR-A/IR-B ratio and IR-A also binds with high affinity IGF2, which signals preferentially through MAPK for mitogenesis and whose basal levels are higher than those of insulin. IGF2 may play a very important role in diabetes and cancer because is part of a very complex transcriptional unit induced by insulin, that contains in addition to IGF2, IGF-binding proteins, degrading enzymes important for extracellular matrix remodeling and migration and long noncoding and micro RNAs, which coordinately control systemic metabolism and IR expression. This complexity, revised by Refs. [ , ], hampers the understanding of the role of IGF2 in the interplay between diabetes and cancer. IGF1 is also induced by insulin, binds IGF1R and signals predominantly through MAPK for mitogenesis. Insulin may bind its own receptor and with less affinity (perhaps, through exogenous administration of pharmacological doses) related receptors such as IGF1 receptor (IGF-1R). In addition, hybrid receptors containing subunits from IR and from IGF1R, which tend to signal predominantly through MAPK, are increased in cancer cells. Insulin has been suggested to signal predominantly though MAPK for mitogenesis and through PI3K for survival in both, insulin resistance at diabetes and also in cancer cells [ ].
Exogenous insulin induced tumor foci in mouse models for cancer [ ] and induced proliferation in cells from patients or in animals. However, the proliferative effects could not be unequivocally attributed to high levels of insulin due to the coexistence of high circulating levels of leptin or glucose. Insulin effects in animal models are context-dependent [ ].
Leptin/adiponectin proportions are also altered in T2D with leptin signaling through its receptor (ob-R) that belongs to the family of receptors associated to cytosolic tyrosine kinases. ob-R signals through PI3K/GSK3β and MAPK for proliferation, through JAK/STAT and JNK for survival and through Src/MAPK for migration [ ]. In addition, hyperleptinemia in T2D correlates with increased circulating inflammatory cytokines, and a cross-regulation leptin-inflammatory cytokines has been suggested [ ]. Increased leptin and inflammation correlate with growth of neoplasms, but hyperleptinemia in animal models does not alter cancer initiation [ ]. Contrary to leptin, adiponectin levels decreased in both T2D and cancers. Adiponectin signals through its seven transmembrane receptors AdipoR1 and 2 to stimulate AMPK that mediates its anti-inflammatory and antigrowth (mTORC1-driven) effects and through JAK/STAT and p217P27 to slow down the cell cycle and allow apoptosis [ ]. Despite all these evidences, adiponectin effects on cancer initiation/progression vary with animal models and critically depend on the hormonal, cytokine and metabolic context [ , ] as for insulin.
3.3
Immune defects
T2D develops from obesity and is characterized by low grade, diffuse, chronic inflammation. Chronicity results from failure of the immune mechanisms to resolve inflammation, slows down tissue healing and alters the inflammation-dependent modulation of the innate and adaptive immune responses. In T2D, high levels of circulating proinflammatory cytokines [ ] and the presence of more frequent and longer infections [ ] reflect a status of immune compromise that may favor immune escape for cancer initiation, since immune surveillance is a critical barrier for cancer development. Longer infections may additionally obey to the growth advantage offered for microorganisms by the glucose-rich plasma of the diabetic patient. However, high levels of plasma glucose and lipids induce changes in the functionality of humoral and cellular components of the innate and adaptive immune responses, suggesting the existence and contribution of immune defects in T2D. Hyperglycemia induces nonenzymatic addition of glucose to other molecules rendering advanced glycation end products or AGEs. Humoral components of the innate response such as protein C reactive (PCR) or complement components become glycated and lack functionality. At the cellular compartment of the innate response, protein and lipid glycation diminish the exhibition of class I major histocompatibility complex, leading to antigen presentation defects that impair immune surveillance. Hyperglycemia regulates natural killer (NK) receptors that mediate apoptosis and interfere with cytotoxic NK cell activation, leading to increased NKs but dysfunctional. Humoral components of the adaptive response, antibodies, also suffer glycation that compromises their ability to fix the complement, reduces their levels, and compromises class switch [ ]. Additionally, cellular components (number, proportions and activation) of the adaptive response become unbalanced by the metabolic environment in diabetes, cooperating as well with the increased circulating proinflammatory cytokines [ ]. Conversely, immune cells were shown to contribute to the regulation of insulin sensitivity and systemic metabolism, so their imbalances will add to the loss of glucose homeostasis. Altogether, the metabolic environment of diabetes alters the levels and activity of immune humoral molecules, and the numbers and activity of immune cell subsets causing imbalances that hamper regulation. Changes imposed by diabetes in immune components affect circulating immune cells but also infiltrates in white adipose tissue (WAT) or in tumors [ ]. Profiling immune infiltrates is difficult due to the dynamism of the cytokine environment, able to induce transdifferentiation (for example between subsets of innate lymphocyte cells or ILCs), and leading to changes in the output of cytokine signaling.
WAT immune infiltrates, innate and adaptive components, differ in diabetic individuals. Stressed adipocytes (from obesity) secrete proinflammatory cytokines, free fatty acids and adipokines. Proinflammatory cytokines unbalance innate cell subsets, especially among macrophages and ILCs. Macrophages polarize toward proinflammatory phenotype (M1) causing an imbalance M1/M2 [ ]; ILCs comprises ILC1 to ILC3 subsets, where ILC1s are increased and ILC2s decreased in the WAT of T2D subjects and in the circulation of many patients with solid tumors, suggesting a predisposition in T2D patients [ , ]. ILC1s are defined by the production of inflammatory cytokines: TNFα, IFNγ, IL8, and IL10 and include NK cells, which appear increased but dysfunctional. ILC2s produce IL4, IL5, and IL13 [ ]. The adaptive WAT infiltrates in T2D also show T-cell subset imbalances. CD4+ lymphocytes, including Treg and T-helper (Th1, Th2 and Th17) show decreased Treg and Th2 and increased Th1 and Th17 [ , ]. Decreased Treg leads to increased CD8+, generally involved in macrophage recruitment and insulin resistance. Likewise, a specific CD8+ subset, mucosa-associated T cells or MAIT that produces IL17, is specifically enriched in the WAT by T2D and in solid tumor infiltrates through their evolution [ ].
Similar imbalances in innate cells (macrophages, ILCs, antigen presenting, dendritic cells) and adaptive cells (CD4+ subset imbalances and increased dysfunctional CD8+ subsets) have been found in circulation in patients with solid tumors [ ]. Thus, proinflammatory cytokine production by stressed adipocytes in T2D induces M1 macrophage polarization, producing more inflammatory cytokines that initiate a feed forwards cycle. Cytokines produced act on epithelial, immune, stromal, tumor, and other cells inducing changes in proliferation, invasiveness, tumor cell production of cytokines, and glucose transport and metabolism at many levels [ ]. This contributes to change the metabolic environment, which will in turn promote dysfunction in humoral and cellular components from the innate (ILCs) and adaptive (T cytotoxic) response.
Cytokines signal through JAK/STAT, MAPK, PI3K, and AMPK. All these pathways regulate and are strongly regulated by metabolism, reflecting the reciprocal and very dynamic control between inflammatory cytokines and metabolism [ ].
Tumor infiltration by innate and adaptive cells evolves with tumor progression, and there is increasing attention to profile their evolution. Tumor-infiltrated lymphocytes (TILs) and tumor associated macrophages (TAMs) profiles have been correlated with tumor prognosis. Low CD8+ TIL and high Treg at the invasive front correlated with worse prognosis (lower OS and DFS) in colorectal cancers [ ]. Likewise, for advanced cancers, MAIT cells decreased in circulation and increased at the tumor infiltrate [ ]. For the humoral counterpart, a cytokine profile enriched in Il-17 from TILs and IL-6, TNFα, TGFβ, and VEGF from TAMs correlated with tumor progression. Thus, inflammation may cooperate with metabolic alterations in diabetes to drive tumor initiation and progression [ , ]. Cytokines and glucose metabolism cooperate to increase ROS and reactive nitrogen species (RNS) levels causing DNA damage and mutations. Metabolic adaptations drive the production of oncometabolites that alter chromosome and genome stability. Cytokines and lipids induce ER stress that triggers signals for phenotype switch and cancer cell diversification [ ].
3.4
Microbiome
An additional defense of our organism comes with the microbiota, composed by symbiotic fungi, protists, and bacteria, that feed from nutrients discarded by our body and produce substances that finely tune our systemic metabolism and immune responses. Microbiota is enriched in the intestine as microbiota intestinalis (MI), where aberrant interactions with the host are prevented by the gut barrier (epithelial cells joined through tight junctions and covered by mucin), avoiding immune responses against them. The microbiome composition is remodeled by the environment and not surprisingly is altered in T2D subjects, human and in mice, due to gut barrier damages caused by hyperglycemia [ ]. This exposes MI antigens and lipopolysaccharide to the toll-like receptors (TLRs) exhibited by neighbor mucosa and myeloid cells worsening inflammation. Barrier damage allows the growth of opportunistic bacteria further damaging the barrier, competing by the niche to reduce MI diversity and producing genotoxins able to cause genetic lesions or signal through cancer cell signaling pathways, especially those related to beta-catenin [ , ]. Damaged barrier cooperates with and enhances immune response defects imposed by T2D and described above, to allow opportunistic growth further altering MI composition.
The composition of stool microbiota reflects MI changes in T2D and are characterized by decreased butyrate-producing bacteria, altered Firmicutes/Bacteroides proportions [ , ] and increased Fusobacterium , especially F . nucleatum , which has been reported to increase beta-catenin-related cancer cell signaling through the production of adhesins, reduction of E-cadherin, and inhibition of NK cells [ ]. In T2D patients, the MI is also enriched in Bacteroides fragilis , producer of the toxin fragilisin , able to break E-cadherin to disrupt the gut barrier and increase inflammation. Both inflammation and E-cadherin destruction favor cancer cell signaling.
Altogether, changes in metabolism, hormonal signaling, immune defects, and microbiome alterations, present in T2D and cancer, influence each other and the progression of both diseases, see Fig. 15.2 , and this need to be considered in the treatment of patients with T2D and cancers.
4
How can knowledge on diabetes-cancer associations improve the management of patients with diabetes and cancer together?
If management of diabetes is a challenge in terms of the lifestyle of patients, cancer diagnosis will come as a defiance. The variability of diabetes, cancer, and their treatments hinders the development of specific guidelines to treat patients that suffer both diseases and challenges personalized medicine. Diabetes poses on cancer problems of metabolism (lack of glycemic and lipidemic controls), signaling (altered insulin pathways) immunity and drug interference that deserve special attention, but the treatments for each disease represent yet another level of complexity for careful consideration, Fig. 15.2 .
4.1
Treatment interference
Drugs directed to control diabetes may interfere either with cancer biology or with cancer treatments, Fig. 15.2 . Conversely, cancer (and its treatments) threats diabetes management due to frequent induction of vomiting, anorexia, and slow down intestinal movements that retard nutrient absorption. Antineoplastic drugs may cause or aggravate diabetes and many classical antineoplastic treatments, glucocorticoids, and drugs targeting PI3K/Akt/mTOR (PAM inhibitors) lead to hyperglycemia [ ] and/or hyperlipidemia [ ] that worsen the course of diabetes and hamper the adjustment of antidiabetics, Fig. 15.2 . Likewise, checkpoint inhibitors that target PD1/PD-L1 signaling pathways can cause T1D [ ]. Therefore, drug interference between antidiabetic and antineoplastic treatments is an important issue to be considered and still awaits careful prospective studies.
4.2
Metabolic control
Hyperlipidemia and specially elevated LDL-cholesterol awithre associated to increased risk of certain cancers such as colorectal in T2D patients [ ], and some guidelines to carefully monitor it have been reported [ ]. Cholesterol levels are reduced with statins, suggesting that their beneficial effect may extend from T2D to cancer patients. But many statins are degraded at the liver by CYP enzymes and compete with antineoplastic drugs for degradation, misadjusting the doses of antineoplastic. This may explain controversial epidemiological results concerning distinct statins in some cancers [ ].
Lack of glycemic control in diabetic patients with cancer is perhaps the most common stumbling block. Hyperglycaemia poses a clear challenge for surgical interventions due to more frequent and longer infections, aggravates inflammation, and correlates with bad prognosis. Hypoglycemia may develop in cancer patients, who often experience vomiting, diarrhea, anorexia, and malabsorption as a consequence of cancer itself or of suboptimal adjustment of antidiabetic or antineoplastic treatments due to drug interferences. Hypoglycemia, when it remains untreated, may quickly lead to life-threatening effects, whereas hyperglycemia damages may be acute (polyuria, glucosuria, dehydration, and ketoacidosis) or long-term developed, damaging vessels and vital organs [ ]. Hyperglycemia-induced damages to the gut barrier cause dysbiosis, increasing inflammation and damaging the immune response, thus contributing to cancer onset. In the cancer context, hyperglycemia feeds cancer cell proliferation and lipid uptake, that may direct phenotype switch for cancer cell diversification [ ]. Thus, since both hypo- and hyperglycemia increase morbidity and mortality, glycemic control becomes critical in patients with diabetes and cancer. However, the glycemic targets should be flexible, and according to the cancer prognosi,; glycemia values might be preferred over glycated hemoglobin, given the frequent hemoglobin depletion in cancer.
Patient management will depend on (i) the site and type of cancer involved which will determine anticancer therapy, (ii) type of diabetes and its treatment, and (iii) the timing for diabetes and cancer.
The site and type of cancer determine its association with diabetes and the type of therapy to use, surgery, radiation, chemotherapy, immunotherapy, hormone therapy, or combinations, all of which may worsen diabetes and/or interfere with antidiabetic drugs. Most anticancer therapies alter glycemic control in diabetic, and sometimes in nondiabetic patients [ ]. This includes classical platinum derivatives, microtubule organizers, etc. and also glucocorticoids (GCs), PAM inhibitors, and checkpoint inhibitors. GCs, used as coadjuvant or to palliate adverse effects of antineoplastic agents, increase insulin resistance and liver gluconeogenesis [ ]. PAM inhibitors induce hyperglycemia and hyperlipidemia [ , ]. Checkpoint inhibitors trigger beta pancreatic cell destruction in 1% patients inducing acute and severe hyperglycemia [ ].
For the type of diabetes, management of T1D requires insulin and is generally simpler than T2D, even in the context of a coexisting cancer. T1D patients do not usually show insulin resistance and exogenous insulin should be adjusted. The simpler procedure is adjustment to the patient status and response to treatments, of the bolus of rapid-acting insulin administered before meals. Rapid-acting insulin should always be adjusted before PET scans. Basal, long-acting insulin might need modification pre- and postsurgery to avoid hypoglycemia. Pumps might be disconnected for surgical interventions and their settings should be adjusted according to medication and considering the pharmacokinetics, especially if glucocorticoids are used. Specific guidelines have been reported to avoid increased mortality for T1D patients with cancer [ ]. However, most diabetic patients suffer T2D, carry pre-existing insulin resistance and are subjected to wide diversity of treatments which complicates management. T2D patients may suffer hypoglycemia (especially those treated with insulin or secretagogues like sulfonylureas) and hyperglycemia episodes [ ]. Many T2D patients are treated with insulin or insulin-secretagogues, which will compete with widely used antineoplastic treatments of the classes of tyrosine kinase inhibitors or PAM inhibitors and will need careful dose adjustments to compensate for drug interference. Treating T2D patients with insulin sensitizers such as metformin or pioglitazone may counteract the insulin resistance induced or aggravated by glucocorticoids and antineoplastic drugs. Metformin was suggested to be beneficial for some cancers such as colorectal even without diabetes but results from clinical trials are not yet conclusive. Nonetheless, metformin doses should be graduated carefully since maladjustment may cause ketoacidosis. Regarding pioglitazone, its association to increased risk of bladder cancer [ ] would make advisable to avoid it, especially in patients with familial antecedents of bladder cancer until more data are available. Alternatively, the novel sodium-glucose transporter 2 inhibitors (SGLT2-I) may offer an interesting option in T2D patients with cancer based on their antiproliferative effects in some cancers.
Concerning the timing of diabetes, both the disease and antidiabetic treatments usually become more complex with the duration of diabetes. Normally, the longer duration of diabetes, the more metabolic and immune derangements develop to potentially interfere with cancer and its treatments. In terms of cancer timing, management of initial stages of cancer (with long survival prognosis) requires a consideration of long-term consequences of hyperglycemia such as infections, hypercoagulability, osmotic diuresis, weight loss, and ketoacidosis. In contrast, in advanced cancers (with shorter survival, i.e., less than 5 years), preserving the quality of life must be prioritized, i.e., preventing dehydration due to acute hyperglycemia effects (ketoacidosis and hyperosmolarity).
Therefore, management of patients with diabetes and cancer requires an effort to understand the mechanisms of action of drugs used to treat both cancer and diabetes and their possible interference, Fig. 15.2 , and a very close and careful control of glycemia and lipidemia, as well as preparing rescue strategies to quickly restore glycemia and lipidemia.
4.3
Diet modulation may be used to impact the microbiota and the immune response
Unbalanced diets enriched in fat and sucrose (high-fat diets or HFD) are associated to obesity and T2D development in humans and used in animal models to induce diabetes. HFDs alter both the microbiome and the immune response. HFDs strongly reduce MI diversity since their saturated fatty acids (SFAs) enhance the growth of LPS-producing bacteria that interact with TLRs in cells of the mucosa, inducing NFk B and leading to inflammation. HFDs also increase the production by the MI of secondary bile acids, which disrupt beta-catenin/E-cadherin interactions at the membrane of intestinal epithelial cells to promote beta-catenin-dependent cancer cell signaling [ ]. Concerning the immune response, HFD strongly reduces in mice the most abundant antibody in mucosae: IgA [ ]. Since IgA is critical to induce host tolerance to its symbiotic microbiota, IgA reduction in T2D contributes to MI dysbiosis. Thus, avoidance of HFDs makes logic to prevent diabetes and with it the rise in certain cancers incidence.
In contrast to HFD, fiber-rich diets are metabolized by MI to produce short chain fatty acids (SCFAs) such as propionate, butyrate, or acetate, which are differentially metabolized by healthy or cancer cells. Healthy cells obtain energy from SCFAs, whereas in cancer cells SCFAs inhibit deacetylases (HDACs), bind specific G-protein coupled receptors to induce the inflammasome and promote lysosome and mitochondria membrane alterations that increase ROS production and apoptosis, with a net beneficial effect. Beneficial effects promoted by SCFAs produced by MI include propionate-enhanced intestinal-brain signaling to increase satiety, improved insulin sensitivity and glucose tolerance [ , ]; acetate induced antiproliferative effects; butyrate interference with proliferation, invasiveness and inflammation, mediated through reduced cytokine production in cancer cells, and modulation of TLR4 to prevent cancer development. Butyrate also regulates the balance between two critical T-cell CD4+ subsets with opposite effects on inflammation: increasing suppressor Treg and decreasing proinflammatory Th17. Thus, depletion of butyrate-producing bacteria in T2D contributes to inflammation and fiber-rich diets may help in prevention, revised in Ref. [ ].
5
Future perspectives
Knowledge of the molecular mechanisms that mediate cancer initiation/progression or drug interferences in diabetic patients will provide tools to fight its devastating effects in patients with diabetes and cancer and may help to understand each disease separately. Also, definition of the immune defects imposed by diabetes on circulating and infiltrated immune components may help in early cancer detection and treatment.
Many questions remain to be understood and their answers should improve the wellbeing of diabetic patients. For example: What is different in diabetic patients that do not develop cancer? Is there a crucial window of opportunity in the first 5 years of diabetes to detect and/or interfere with cancer development? Is it possible to quantify the decreased survival in diabetic-cancer patients for each cancer site compared to nondiabetic patients to guide future management? Does diabetes increase cancer initiation or cancer progression? Do diabetic patients get worse overall survival because they have more metastasis, that is, does diabetes favor metastasis? If there are antidiabetic treatments that promote and others that oppose specific associated cancers, Does this inform on individual factors contributing to cancer from diabetes? Do these results guide diabetic patient management according to family history? If some antineoplastic treatments induce diabetes, could it be prevented? Are there enough well-controlled studies and/or clinical trials? Solving these questions will help to improve management guidelines for diabetic patients.
As there are no specific guidelines for the treatment of patients with diabetes and cancer, specific screening protocols for early detection of the associated cancers in diabetic patients are also missing. It would be advisable to develop screening policies, at least for cancers of high incidence, poor prognosis, and clear association to diabetes such as colorectal cancer.
References
Related posts:
 Overview on chronic complications of diabetes mellitus
Overview on chronic complications of diabetes mellitus
 Molecular mechanisms involved in hyperglycemia-induced cellular damage
Molecular mechanisms involved in hyperglycemia-induced cellular damage
 Diabetic retinopathy: New insights
Diabetic retinopathy: New insights
 Diabetic neuropathy: Current issues in diagnosis and prevention
Diabetic neuropathy: Current issues in diagnosis and prevention
 Diabetes-related cognitive impairment and dementia
Diabetes-related cognitive impairment and dementia
 Role of glycemic legacy and glycemic variability in the development and progression of complications of diabetes
Role of glycemic legacy and glycemic variability in the development and progression of complications of diabetes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


