Better understanding of the underlying principles of tumor biology and immunology, enhanced by recent insights into the mechanisms of immune recognition, regulation, and tumor escape has provided new approaches for cancer immunotherapy. This article reviews the current status and future directions of cancer immunotherapy, with a focus on the recent encouraging results from immune-modulating antibodies and adoptive cell therapy.
Key points
- •
Despite recognition of durable therapeutic responses in some patients with cancer historically, immunotherapeutic agents have provided limited evidence of clinical success until recently.
- •
Recent discoveries by which tumors evade immune T-cell recognition have led to a variety of clinically effective cancer immunotherapies.
- •
Understanding the pivotal role of the tumor microenvironment in suppressing antitumor immunity and the exploration of combinatorial treatment regimens will further enhance the efficacy of cancer immunotherapies.
History of cancer immunotherapy
The first systematic study of immunotherapy for the treatment of malignant tumors was conducted by a New York bone sarcoma surgeon, William B. Coley (1862–1936), whose line of clinical research began in the 1890s. Coley’s interest in immunotherapy stemmed from the loss of a young patient with a soft tissue sarcoma of the arm, who developed metastatic disease and died shortly after Coley’s radical excision of the tumor. Coley investigated the medical records of the hospital and found a patient with a four-time recurrent inoperable sarcoma of the neck, which regressed after a postoperative infection of erysipelas (a superficial, streptococcal infection of the skin). This incident stimulated Coley’s clinical research, and he started to infect patients with cancer with various bacterial isolates. He developed “Coley’s toxin,” the combination of heat-killed Streptococcus pyogenes and Serratia marcescens . He started intratumoral injections of Coley’s toxin in 1891, and treated hundreds of patients with inoperable bone and soft tissue sarcomas using immunotherapy over the ensuing 40 years. Despite a remarkable result of durable response (5-year survival rate >44%) in selected patients with inoperable soft tissue sarcoma, his strategy gradually fell out of favor because of poorly controlled and documented patient follow-up and many different preparations and administration of the toxins making it difficult for others to reproduce his results. In addition, Coley’s successes lacked a solid theoretical foundation. Clinical interest in cancer immunotherapy diminished in preference to the more broadly applicable chemotherapy and radiation treatment. However, Coley’s intriguing observations and encouraging clinical results led to the understanding that the immunologic host response may affect the biologic behavior of malignant tumors and that manipulation of the immune system might cause recognition of the tumor, initiation of tumor-specific immunity, and eventually tumor destruction.
History of cancer immunotherapy
The first systematic study of immunotherapy for the treatment of malignant tumors was conducted by a New York bone sarcoma surgeon, William B. Coley (1862–1936), whose line of clinical research began in the 1890s. Coley’s interest in immunotherapy stemmed from the loss of a young patient with a soft tissue sarcoma of the arm, who developed metastatic disease and died shortly after Coley’s radical excision of the tumor. Coley investigated the medical records of the hospital and found a patient with a four-time recurrent inoperable sarcoma of the neck, which regressed after a postoperative infection of erysipelas (a superficial, streptococcal infection of the skin). This incident stimulated Coley’s clinical research, and he started to infect patients with cancer with various bacterial isolates. He developed “Coley’s toxin,” the combination of heat-killed Streptococcus pyogenes and Serratia marcescens . He started intratumoral injections of Coley’s toxin in 1891, and treated hundreds of patients with inoperable bone and soft tissue sarcomas using immunotherapy over the ensuing 40 years. Despite a remarkable result of durable response (5-year survival rate >44%) in selected patients with inoperable soft tissue sarcoma, his strategy gradually fell out of favor because of poorly controlled and documented patient follow-up and many different preparations and administration of the toxins making it difficult for others to reproduce his results. In addition, Coley’s successes lacked a solid theoretical foundation. Clinical interest in cancer immunotherapy diminished in preference to the more broadly applicable chemotherapy and radiation treatment. However, Coley’s intriguing observations and encouraging clinical results led to the understanding that the immunologic host response may affect the biologic behavior of malignant tumors and that manipulation of the immune system might cause recognition of the tumor, initiation of tumor-specific immunity, and eventually tumor destruction.
Classification of cancer immunotherapy
Cancer immunotherapy can be classified into two types: active and passive ( Fig. 1 ). Active immunotherapies stimulate the endogenous immune system, and are further divided into two systems: specific and nonspecific immunotherapy. Active specific immunotherapy includes either prophylactic or therapeutic approaches. Prophylactic vaccines target infectious agents that cause or contribute to the development of cancer, which include hepatitis B virus (HBV) vaccine and human papillomavirus (HPV) vaccine. Therapeutic vaccines are designed to treat existing cancers and may use attenuated whole cancer cells, tumor lysates, or purified tumor antigens, with or without adjuvants, to stimulate the patient’s immune system. Active nonspecific immunotherapy uses the components of the nonspecific immune system of the body. These components include immune adjuvants, such as bacillus Calmette-Guérin (BCG), and cytokines including interleukin (IL)-2, IL-12, granulocyte-macrophage colony–stimulating factor (GM-CSF), tumor necrosis factor (TNF)-α, and interferon (IFN)-α-2b.
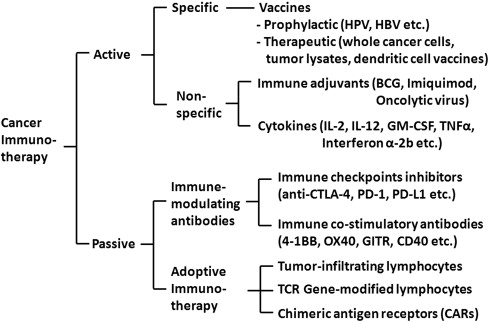
Passive immunotherapies do not stimulate the immune system and bypass the requirement to activate endogenous immunity. These therapies are comprised of antibodies or cells that are made or modified in the laboratory and administered to patients to provide or enhance immunity against cancer. The passive transfer of monoclonal antibodies (mAbs) and cells to augment antitumor immunity are effective treatments for a variety of hematologic and solid malignancies.
Examples of mAbs used to enhance the immune system include anti–cytotoxic T-lymphocyte antigen (CTLA)-4, anti–programmed death (PD)-1, anti-PD ligand 1 (PD-L1), and anti–4-1BB. Adoptive cell therapy (ACT) is another form of passive immunotherapy that involves identification, isolation, activation, and reinfusion of autologous lymphocytes reactive to the tumor into patients.
Prophylactic and therapeutic cancer vaccines
Prophylactic vaccines have been used with considerable success for the prevention of cancers attributable to infectious agents, such as HBV and HPV. HBV vaccines protect against HBV infection, and the potentially subsequent severe and deadly sequelae, such as cirrhosis and hepatocellular carcinoma. The quadrivalent HPV vaccine is efficacious in preventing persistent cervical infection with HPV-6, -11, -16, or -18 and high-grade cervical intraepithelial neoplasia associated with these infections. Recently, the quadrivalent HPV vaccine has been shown to also prevent vaginal, vulvar, and anal precancers.
In contrast, the development of therapeutic vaccines to treat established tumor has been problematic. Therapeutic cancer vaccines directly target not the tumor, but the immune system, which in turn targets the tumor and its microenvironment to recalibrate the existing host-tumor interaction, skewing the balance from tumor tolerance toward tumor control. Although therapeutic cancer vaccines, when used as monotherapy, have minimal toxicity compared with conventional chemotherapy, the efficacy of ex vivo derived therapeutic vaccines has still to be proved for human cancers, and the limited success has been attributed to a variety of factors regarding the preparation and administration of the vaccine, the disease stage of patients in clinical trials, or the heterogeneous nature of malignant tumors.
The first therapeutic cancer vaccine approved by the US Food and Drug Administration is sipuleucel-T to treat minimally symptomatic or asymptomatic metastatic castration-resistant prostate cancer. Sipuleucel-T is a cellular product comprised of autologous dendritic cells pulsed ex vivo with a recombinant fusion protein (PA2024) consisting of GM-CSF and prostatic acid phosphatase. Dendritic cells are potent antigen-presenting cells that are responsible for inducing T and B immune responses. After processing, the dendritic cells are reinfused into the patient, with the goal of generating an immune response against prostatic acid phosphatase. A phase III randomized placebo-controlled double-blind multicenter study enrolled 512 men with minimally symptomatic or asymptomatic metastatic castration-resistant prostate cancer and randomized them 2:1 to receive sipuleucel-T or placebo every 2 weeks for three treatments. Sipuleucel-T was found to be well tolerated and prolonged median overall survival by 4.1 months compared with placebo (25.8 vs 21.7 months, respectively) but failed to show benefit in progression-free survival, and tumor regressions were rare.
Immune adjuvants
Cancer cells often express a variety of abnormal proteins (tumor antigens) as targets for an immune response. Because tumors are poorly immunogenic, spontaneous immune responses to these antigens are weak and rarely sufficient to cause tumor regression. However, the local administration of immune-activating agents (immune adjuvants) can increase the strength of the immune response and induce protective immunity. In general, immune adjuvant-based therapies have only proved effective against early stage tumors.
In 1908, Albert Calmette and Camille Guérin of the Pasteur Institute in Lille, France began to investigate a vaccine for human tuberculosis and developed the attenuated form of bovine tuberculosis, Mycobacterium bovis , or BCG. The use of BCG as a cancer therapy was started when an autopsy series by Raymond Pearl in 1929 showed a lower frequency of cancer in patients with tuberculosis. Since the first report on successful treatment of superficial bladder cancer with intravesical BCG in 1976, it remains a clinically accepted and approved therapy for bladder cancer. Combination therapy with surgery and BCG was associated with progression-free survival in 62% of patients, compared with 37% of patients who underwent surgery alone. Although the mechanism in BCG immunotherapy is not fully understood, the degree of local inflammatory reaction correlates with efficacy, suggesting an immune response elicited by BCG rather than a direct antitumor effect.
Invasion of microbial pathogens triggers immune responses by activating evolutionarily conserved pattern-recognition receptors, such as members of the Toll-like receptor (TLR) family. TLRs comprise the main class of cell-surface pattern-recognition receptors that are expressed on antigen-presenting cells, such as macrophages and dendritic cells. TLR ligation was found to activate the innate and adaptive immune systems, and plays an important role in antitumor immunity. A significant amount of effort has been devoted to exploit the therapeutic potential of TLR agonists. Imiquimod is a low-molecular-weight immune-response modifier that can activate TLR-7 on dendritic cells and induce secretion of various cytokines, such as IFN-α, IFN-γ, TNF-α, IL-1, IL-6, IL-8, IL-10, and IL-12. Imiquimod was initially approved in 1997 for the treatment of external anogenital and perianal warts caused by HPV infection. Because Imiquimod was also found to demonstrate efficacy against low-grade epithelial tumors and precancerous lesions, it is now approved for the treatment of superficial basal cell carcinoma and actinic keratosis, the precursor lesion of squamous cell carcinoma, and is being tested for melanoma and vulvar intraepithelial neoplasia.
Another attractive approach is the use of oncolytic viruses that can exert a direct cytotoxic effect and promote immune-mediated tumor rejection. These features have prompted investigators to further explore oncolytic viruses for cancer immunotherapy. Herpes simplex virus-1 vectors have shown promising efficacy against a wide variety of malignancies including colorectal, head and neck, breast, prostate cancer, melanoma, and glioblastoma. A phase II clinical trial with intratumoral injection of attenuated herpes simplex virus-1 engineered to secrete GM-CSF (OncovexGM-CSF) for the treatment of patients with stage IIIc or IV metastatic melanoma has shown 26% objective response rate including uninjected regional and distant soft tissue and visceral metastatic sites.
Cytokines
Cytokines are proteins secreted by cells of the immune system and can be delivered systemically to activate antitumor immunity. IL-2 was first identified as a T-cell growth factor in 1976, and has been found to be an important mediator of immune function through its effects on the growth, development, and activity of T and B lymphocytes, and natural killer cells. Based on potent, dose-dependent immunomodulatory and antitumor activity shown in preclinical models, clinical studies were conducted. Treatment with high-dose, single-agent IL-2 in patients with metastatic melanoma and renal cell carcinoma demonstrated 15% to 20% objective responses and 7% complete remission in metastatic melanoma or renal cell carcinoma.
Subsequent publications of retrospective analysis with extended follow-up of 270 patients treated with high-dose IL-2 conducted at the National Cancer Institute between 1985 and 1993 showed 16% objective response including 17 (6%) complete responses, and 59% of complete responders remained progression-free at 7 years. Phase II studies evaluating the efficacy of high-dose IL-2 for 259 patients with metastatic renal cell carcinoma treated at the National Cancer Institute showed 20% objective responses including 23 (8.9%) complete responders, of which 19 (83%) remained free of recurrence at follow-up between 24 and 221 months. Although immunotherapy with high-dose IL-2 carries significant risk of serious systemic toxicities, which requires administration as an in-patient, these durable complete responses led to the approval of high-dose IL-2 for clinical use in 1992 for metastatic renal cell carcinoma and in 1998 for metastatic melanoma.
IFNs are pleiotropic cytokines and were first discovered as an antiviral agent during studies on virus interference. IFN-mediated signaling and transcriptional activation of cellular gene expression are best understood in the context of JAK-STAT pathway proteins, which is a major signaling system that cells use to transmit extracellular information from many cytokines and growth factors to the nucleus. Although the antitumor mechanism of action for IFN-α-2b therapy in patients with melanoma has not been universal, preclinical and clinical models suggest that the effects of IFN-α-2b are mediated by host cells rather than direct effects on tumor cells. The use of IFN-α-2b has been studied as an adjuvant treatment in patients with melanoma. High-dose IFN-α-2b demonstrated increased disease-free survival and in some studies, overall survival in high-risk patients with melanoma in randomized clinical trials. A meta-analysis analyzing data from 14 randomized trials including 8122 patients showed adjuvant immunotherapy with IFN-α-2b has been found to prolong disease-free and overall survival in selected patients with melanoma at increased risk for disease dissemination. IFN-α-2b has a large variety of possible side effects in virtually all organ systems, which often hamper the attainment of optimal dose intensity and sometimes even necessitates premature cessation of therapy. Pegylated IFN-α-2b has been developed to decrease the frequency with which IFN-α-2b is administered while maintaining a high level of exposure. A randomized clinical trial that enrolled 1256 patients with resected stage III melanoma showed significantly increased 7-year relapse-free ( P = .06), distant metastasis-free ( P = .02), and overall survival ( P = .006) with pegylated IFN-α-2b in patients with sentinel lymph node involvement and ulcerated melanoma compared with observation.
Immune-modulating antibodies
The capacity of mAbs to induce tumor-directed T-cell responses is intriguing. Intratumoral T-cell composition and distribution have been associated with clinical outcomes, suggesting the relevance of T cells in antitumor immunity. Moreover, T cells can target intracellular antigens that are thought to be inaccessible to antibody therapies. The ultimate amplitude and quality of the response of T cells is regulated by a balance between costimulatory and inhibitory signals (immune checkpoints). After activation, T cells upregulate the expression of inhibitory receptors, which protects against deleterious autoimmunity. The importance of these pathways was highlighted by studies showing that total blockade by genetic disruption led to massive T-cell hyperproliferation and protracted multiorgan autoimmunity. Thus, under normal physiologic conditions, immune checkpoints are crucial for the maintenance of self-tolerance to prevent overactivation. Cancer takes advantage of this ability to hide from the immune system by exploiting a series of immune escape mechanisms that were developed to avoid autoimmunity.
Recently, preclinical and clinical data showed that blockade of these immune checkpoints can significantly enhance antitumor immunity. Blockade of one of these checkpoints, CTLA-4, provided the first evidence of improvement in overall survival for the treatment of patients with metastatic melanoma. CTLA-4 is a key negative regulator, expressed exclusively on T cells, and counteracts the activity of the T-cell costimulatory receptor, CD28 ( Fig. 2 ). After antigen is engaged by T-cell receptor (TCR), CD28 signaling amplifies TCR signaling to activate T cells.
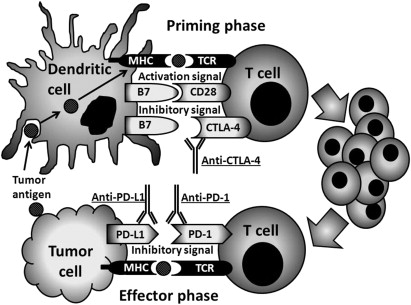
CD28 and CTLA-4 share B7 family of accessory molecules (CD80, CD86) expressed by antigen-presenting cells. Because CTLA-4 has a much higher affinity for CD80 and CD86, ligation to CTLA-4 inhibits further activation and expansion, thereby controlling the progress of an immune response. CTLA-4 ligation is not only through the activated CD4 and CD8 T cells, but also associated with enhancement of regulatory T-cell immunosuppressive activity. In a placebo-controlled phase III trial, 676 patients with relapsed-refractory metastatic melanoma were randomly assigned in a 3:1:1 ratio to ipilimumab plus glycoprotein 100 (gp100) peptide vaccine, ipilimumab alone, or gp100 alone. Overall survival was significantly increased in patients given ipilimumab (22%, 24%, and 14% at 24 months, respectively). Of note, 18% of the ipilimumab-treated patients survived beyond 2 years compared with 5% of patients receiving the gp100 peptide vaccine alone. In another randomized trial involving 502 patients with previously untreated metastatic melanoma, the addition of ipilimumab to standard dacarbazine alone was shown to improve overall survival compared with dacarbazine alone (11.2 months vs 9.1 months; P <.001). Immune-related toxicity involving various tissue sites, such as enterocolitis, hepatitis, dermatitis, and endocrinopathy, were also observed in 25% to 30% of patients. PD-1 is another T-cell coinhibitory receptor with a structure similar to that of CTLA-4 but with a distinct biologic function and ligand specificity. PD-1 predominantly regulates effector T-cell activity within tissue and tumors (effector phase), whereas CTLA-4 predominantly regulates T-cell activation (priming phase) (see Fig. 2 ). PD-1 is more broadly expressed than CTLA-4: it is induced on non–T lymphocytes subsets, including B cells and natural killer cells. PD-1 has two principal ligands, PD-L1 and PD-L2. PD-L1 is not only expressed on many tumors, but also on myeloid cells in the tumor microenvironment.
PD-L2 is expressed on antigen-presenting cells and various tumors, such as B-cell lymphoma and Hodgkin disease. Expression of PD-L1 is correlated with unfavorable prognosis in cancers of lung, kidney, pancreas, and ovaries. Ligation of PD-1, which is expressed on a large proportion of tumor-infiltrating lymphocytes (TILs) from many different tumor types, renders these cells unresponsive or exhausted as has been suggested by decreased cytokine production by PD-1 + compared with PD-1 − TILs from melanomas. Blockade of this interaction by PD-1/PD-L1 axis is expected to revitalize these exhausted tumor-specific T cells. Recently, two large clinical trials of anti–PD-1 therapy or anti–PD-L1 therapy reported impressive durable response rates. Nivolumab is a humanized mAb that targets the PD-1 protein. This mAb was evaluated in a phase I/II study in 296 patients with a variety of heavily pretreated malignancies including melanoma (104 patients); renal cell cancer (34 patients); and non–small cell lung cancer (122 patients). Cumulative response rates were 28% among patients with melanoma, 27% among patients with renal cell cancer, and 18% among patients with non–small cell lung cancer. Of note, none of the patients with PD-L1 − tumors had an objective response, whereas 36% of patients with PD-L1 + tumors had and objective response ( P = .006). BMS-936559 is a mAb that binds to PD-L1, and has been tested in a dose escalation phase I/II study in 207 patients with a variety of malignancies including melanoma (55 patients); renal cell cancer (17 patients); non–small cell lung cancer (75 patients); and ovarian cancer (17 patients). Objective response rate was 17% in melanoma, 11% in renal cell cancer, 10% in non–small-cell lung cancer, and 6% in ovarian cancer. Responses lasted for 1 year or more in 50% of patients with at least 1 year of follow-up. Autoimmune phenomena have been reported with blockade of PD-1/PD-L1 axis, albeit less frequently than with CTLA-4–specific antibodies, which is consistent with the generally milder autoimmune phenotype seen in PD-1 −/− mice compared with CTLA-4 −/− mice.
Another strategy to augmenting T cell–specific immunity against tumor cells aims to activate costimulatory receptors including glucocorticoid-induced TNF receptor, 4-1BB, OX40, and CD40. Glucocorticoid-induced TNF receptor, OX40, and 4-1BB are members of TNF receptor family expressed on T cells, and ligation of these receptors augments effector T-cell function. Agonistic antibodies directed against each of these proteins were found to increase the efficacy of antitumor immunity in preclinical model.
CD40 is a member of the TNF super family and costimulatory molecule that is expressed by various immune cells and by cancer cells of different histologies. CD40 expression on immune cells has been implicated in the regulation of humoral and cellular immunity, whereas CD40 expression on certain tumor cell types has been implicated in proapoptotic and antiproliferative activity. CD40 is also broadly expressed on dendritic cells and its activation by CD40 ligand, found on activated T cells, seems to ‘‘license’’ the antigen-presenting cell for T-cell activation. Consistent with these findings, a recent phase I trial of the anti-CD40 antibody showed encouraging preliminary results in patients with advanced melanoma.
To achieve the desired antitumor immunity while maintaining immunologic tolerance to self-antigens expressed on normal tissue cells to avoid autoimmune response is a major challenge. Despite some encouraging results with anti–CTLA-4 or anti–PD-1/PD-L1 mAb, clinicians need to be aware that targeting the immunologic synapse can backfire. Early clinical trial of an agonist antibody against the costimulatory receptor CD28 caused a massive cytokine storm and multiorgan failure in six healthy volunteers. One of the clinical trials with agonists of another costimulatory receptor, 4-1BB, which are expressed on activated T cells and natural killer cells, was recently suspended because a considerable degree of liver toxicity was observed with higher doses of the antibody. These findings emphasize that the balance between antitumor effects and autoimmune side effects must be considered in clinical targeting of the immunologic synapse.
Adoptive cell therapy
Adoptive T-cell therapy involves the transfer of ex vivo expanded effector cells as a means of augmenting the antitumor immune response. Tumor-specific T cells can be derived from the patient’s peripheral blood, the tumor environment (TILs), or they can be genetically modified to express a high-affinity antitumor TCR. There are several advantages to the use of lymphocyte transfer as an immune-based approach to treat cancer. T cells are potent effectors of adaptive immunity because of their ability to initiate killing after their receptor-mediated engagement of antigens expressed on the surface of tumor cells. T-cell cytotoxicity requires direct contact with target cells, thereby limiting any possible damage to bystander cells. The attractiveness of target-specific approaches lies in avoidance of the serious side effects of other conventional treatments, such as chemotherapy and radiation, which have relatively nonspecific mechanisms of action. A unique feature of the immune response, unlike conventional cancer therapies, is that it can elicit long-term protection from recurring disease. Another significant advantage of T cell-based immunotherapies is that they do not depend on knowledge of tumor location. T cells, through the expression of trafficking molecules, are equipped to search out and destroy widely disseminated heterogeneous tumor cell targets.
In ACT, T cells can be selected in vitro for high reactivity against tumor antigens and grown under conditions that overcome the tolerizing influences in tumor microenvironment. Recently, ACT with TILs has emerged as one of the most effective treatments in patients with metastatic melanoma. TILs from resected melanoma deposits can be grown to large numbers in IL-2 while retaining reactivity against endogenous tumor-associated antigens. This approach was first described in 1988 by Rosenberg and colleagues, but significant improvement in efficacy came in 2002 with the introduction of a nonmyeloablative lymphodepleting chemotherapy preparative regimen given before the adoptive transfer, which could improve enhanced persistence of the transferred T cells. This regimen can be accomplished using chemotherapy alone or in combination with total-body irradiation for the temporary ablation of the immune system in a patient with cancer. Two potential mechanisms enhancing in vivo clonal expansion of administered autologous TILs have been proposed based on studies in preclinical models: the elimination of suppressor T cells ; or the decreased competition by endogenous lymphocytes for homeostatic regulatory cytokines, such as IL-7 or IL-15. In their most recent report, they treated 93 patients with metastatic melanoma refractory to all other treatments with the adoptive transfer of autologous TILs administered in conjunction with systemic IL-2 administration after a lymphodepleting regimens (chemotherapy alone or with 2 or 12 Gy total body irradiation). Objective tumor regression was observed in 49% to 72% of patients (including 20 patients [22%] of complete regression) with the greater the degree of host lymphodepletion, the more effective was the treatment. Responses were durable (>5 years) and 19 of 20 patients have ongoing complete regressions with a minimum follow-up of 57 months.
There are some drawbacks of this approach in addition to the costs and labor intensity of the treatment. First, not all patients have preexisting tumor-reactive lymphocytes that can be expanded ex vivo: only approximately half of melanomas give rise to antitumor TILs and other types of tumor only rarely contain identifiable tumor-reactive lymphocytes. Isolation and expansion of TILs can take 5 to 6 weeks making this treatment difficult for patients with progressive disease. It involves a rather toxic regimen that entails systemic IL-2 administration and preparative lymphodepleting protocol with chemotherapy with or without total body irradiation, making only patients with good performance status eligible for this treatment.
Genetic engineering of circulating autologous lymphocytes from patients with cancer by using genes that encode receptors capable of recognizing cancer antigens to generate high-affinity T cells with specificity for tumors is used to overcome these limitations. These high-affinity T cells can be equipped with conventional TCRs expressing a heterodimeric αβ receptor, which recognize processed antigenic peptides presented by major histocompatibility complex (MHC) proteins or with chimeric antigen receptors (CARs), which recognize tumor-associated antigen through single-chain variable fragments that are isolated from antigen-specific mAbs. Gene encoding TCRs can be obtained from high-affinity T cells isolated from a patient who had an excellent response to ACT and cloned into a retrovirus or lentivirus, which can then be used to transduce autologous T cells from other patients with matching HLA restriction elements. Morgan and colleagues reported the first successful application of gene-modified T cells to treat 15 patients with melanoma with a TCR that targeted the MART-1 melanocyte/melanoma differentiation antigen and achieved an objective response in 2 of 15 patients including one complete response. Since then, this approach has been applied with a variety of antigens including MART-1 and gp100 (melanoma); NY-ESO-1 (epithelial tumor and sarcomas); CEA (colorectal cancer); and 2G-1 (renal cell carcinoma). However, this approach is complicated by potential “on-target, off-organ” adverse effects caused by recognition of low-level expression of tumor-associated antigen on healthy tissues, which can be recognized by high-affinity TCRs. These side effects include vitiligo, uveitis, and auditory toxicity (MART-1 and gp100) and inflammatory colitis (CEA). The major challenge for immunotherapy using gene-modified T cells remains the identification of antigens that can be targeted to destroy the cancer without causing toxicity to normal tissues. The use of CARs that recognize non–MHC-restricted structures on the surface of target cells is highly attractive because tumors can frequently lose tumor antigen expression through the downregulation of MHC expression. In addition, CAR-modified T cells can specifically traffic to tumor sites and persist as memory cells in vivo, compared with mAbs. Currently, the most successful use of CARs has been reported in the treatment of B-cell lymphoma to target the CD19 molecule, which is expressed on most B-cell malignancies and normal B cells. An intriguing report showed regression in a patient with heavily pretreated B-cell lymphoma with anti–CD19-CAR-transduced T cells, and subsequent reports from the same group and others have shown encouraging results. The use of CARs now has broadened the application of ACT to a variety of cancers including renal cell carcinoma, neuroblastoma, colorectal cancer, and ovarian cancer.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


