Cancer Immunotherapy
Steven A. Rosenberg
Paul F. Robbins
Giao Q. Phan
Steven A. Feldman
James N. Kochenderfer
INTRODUCTION
Progress in understanding basic aspects of cellular immunology and tumor-host immune interactions have led to the development of immune-based therapies capable of mediating the rejection of metastatic cancer in humans. Early studies of allografts and transplanted syngeneic tumors in mice demonstrated that it was the cellular arm of the immune response rather than the action of antibodies (humoral immunity) that was responsible for tissue rejection. Thus, studies of immunotherapy have focused on enhancing antitumor immune responses of T cells that recognize cancer antigens. Antibodies that recognize growth factors on the surface of tumors can contribute to tumor regression, primarily by interfering with growth signals rather than by the direct destruction of tumor cells. The use of monoclonal antibodies in cancer treatment will be considered in Chapter 29.
Evidence for specific tumor recognition by cells of the immune system was obtained in experiments first conducted in the 1940s using murine tumors generated or induced by the mutagen methylcholanthrene (MCA). Mice that received a surgical resection of previously inoculated tumors could be protected against a subsequent tumor challenge with the immunizing tumor but not generally protected against challenge with additional MCA tumors. The observation that CD8+ cytotoxic T cells were primarily responsible for mediating the rejection of MCA-induced tumors in mice led to the identification of genes that encoded tumor rejection antigens expressed on murine tumors as well as the subsequent identification of antigens recognized by human tumor-reactive T cells. The identification of widely shared nonmutated tumor antigens led to the expectation that effective vaccine therapies could be developed for the treatment of cancer patients; however, the response rates in clinical cancer vaccine trials targeting these antigens have, to this point, been disappointingly low. Vaccination with viruslike particles expressing human papilloma virus (HPV) proteins are successful in preventing the establishment of cervical cancer and immunization with peptides derived from the oncogenic HPV E6 and E7 proteins can mediate tumor regression in woman with high vulvar neoplasia.1 Immune-based therapies have, however, been identified that mediate the regression of large, established tumor metastases. Nonspecific immune stimulation with interleukin-2 (IL-2) administration can lead to objective clinical responses in patients with melanoma and renal cancer,2 and inhibition of regulatory pathways mediated by CTLA-43 or PD-14 can lead to tumor regression in patients with metastatic melanoma and lung cancer. The adoptive transfer of melanoma reactive T cells can mediate objective clinical responses in 50% to 70% of patients with melanoma,5 and the ability to genetically modify antitumor lymphocytes is expanding this cell transfer therapy approach to the treatment of patients with other cancer histologies.6 Studies aimed at identifying potent tumor rejection antigens, as well as mechanisms that regulate immune responses to cancer, are being actively pursued.
HUMAN TUMOR ANTIGENS
To be recognized by immune lymphocytes, intracellular proteins must be digested and the resulting peptides transported to the cell surface and bound to Class I or II main histocompatibility molecules (Fig. 14.1). A variety of approaches have been used to identify the antigens that are naturally processed and presented on tumor cells. These include evaluating the ability of cells transfected with tumor cDNA library pools along with genes encoding autologous major histocompatibility complex (MHC) molecules, as well as the ability of target cells pulsed with peptides eluted from tumor cell surface MHC molecules for their ability to stimulate tumor reactive T cells. Reverse immunology approaches that involve either repeated in vitro T cell sensitization or in vivo immunization with candidate peptides or proteins have also lead to the identification of tumor antigens. Candidate epitopes identified on the basis of their ability to bind to a particular MHC molecule, however, may not necessarily be naturally processed and presented on the tumor cell surface, and there are conflicting reports on the ability of T cells generated using some candidate epitopes to recognize unmanipulated tumor targets, as discussed further.
Additional tumor antigens have been identified using antisera from cancer patients to screen tumor cell cDNA libraries, a method that has been termed serological analysis of recombinant cDNA expression (SEREX).7 Although some of the proteins identified using this technique are expressed in a tumor-specific manner, many of these antigens are simply expressed at higher levels in tumor cells than in normal cells. This may occur due to the release of normal self-proteins from necrotic and apoptotic tumor cells leading to the generation of antibodies against intracellular proteins that are normally sequestered from the immune system.
Finally, the use of recently described approaches involving whole exomic sequencing of tumor cells has led to the identification of mutated tumor antigens. These studies will be discussed further in the section devoted to mutated tumor antigens
Cancer/Germ-Line Antigens
The first antigen identified as a target of human tumor reactive T cells was isolated by screening a melanoma genomic DNA library with an autologous cytotoxic T lymphocyte (CTL) clone.8 The gene that was isolated, termed MAGE-1, was found to be a nonmutated gene that was a member of a large, previously unidentified gene family, many of whose members encode antigens recognized by tumor reactive T cells.9 Members of this family of antigens are expressed in the testes and placenta, both of which lack an expression of MHC molecules, but often not in other normal tissues, which has led to their designation as cancer germ-line (CG) antigens. Members of the MAGE gene family are expressed in a variety of tumor types, including melanoma, breast, prostate, and esophageal cancers. The expression patterns of three
different cancer/testes antigens in multiple tumor types is shown in Figure 14.2. The NY-ESO-1 antigen—a CG antigen that is unrelated to the MAGE family of genes—is expressed in approximately 30% of breast, prostate, and melanoma tumors, as well as between 70% and 80% of synovial cell sarcomas.10
different cancer/testes antigens in multiple tumor types is shown in Figure 14.2. The NY-ESO-1 antigen—a CG antigen that is unrelated to the MAGE family of genes—is expressed in approximately 30% of breast, prostate, and melanoma tumors, as well as between 70% and 80% of synovial cell sarcomas.10
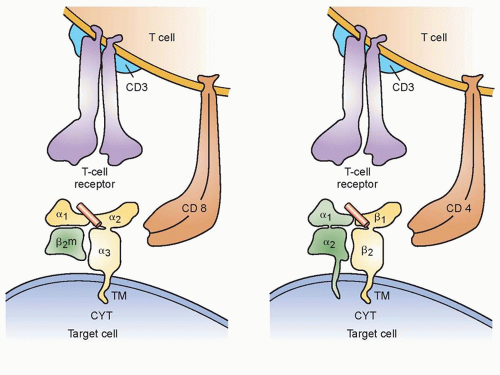 Figure 14.1 CD8 and CD4 cells use different molecules that interact with major histocompatibility complex (MHC) class I and II molecules respectively on the cell surface and serve to potentiate immune reactions. |
Clinical adoptive immunotherapy trials targeting CG antigens have now been conducted in patients with melanoma as well as other tumor types. In a recent trial, objective clinical responses were seen in approximately 50% of patients with melanoma and 80% of patients with synovial cell sarcoma receiving autologous peripheral blood mononuclear cell (PBMC) transduced with a T-cell receptor directed against an HLA-A*02:01 restricted NY-ESO-1 epitope.6 A trial targeting a MAGEA3 epitopes was recently carried out using a T-cell receptor (TCR) isolated from an HLA-A*02:01+ transgenic mouse immunized with the MAGEA3:112-120 peptide.11 Objective clinical responses were observed in five of nine melanoma patients receiving the adoptively transferred PBMC that were transduced with the MAGEA3-reactive TCR.12 Unexpectedly, neural toxicity was observed in three of the patients treated in this trial, two of whom lapsed into a coma and subsequently died. Autopsy samples of patients’ brains revealed that MAGEA12, which encodes a crossreactive epitope recognized by the MAGEA3 TCR, was expressed at low levels in patients’ brains, which may have been responsible for the observed neurologic toxicities. In a recent trial carried out using an affinity-enhanced human TCR directed against the
HLA-A*01:01-restricted, MAGEA3:168-176 epitope, the first two patients receiving TCR-transduced autologous PBMC died of cardiac arrest 4 to 5 days following infusion, which was attributed to cross-reactivity with titin, a protein expressed at high levels in cardimyocytes.13 Taken together, these findings demonstrate the need for caution in evaluating cross-reactivity of high affinity TCRs recognizing tumor antigens.
HLA-A*01:01-restricted, MAGEA3:168-176 epitope, the first two patients receiving TCR-transduced autologous PBMC died of cardiac arrest 4 to 5 days following infusion, which was attributed to cross-reactivity with titin, a protein expressed at high levels in cardimyocytes.13 Taken together, these findings demonstrate the need for caution in evaluating cross-reactivity of high affinity TCRs recognizing tumor antigens.
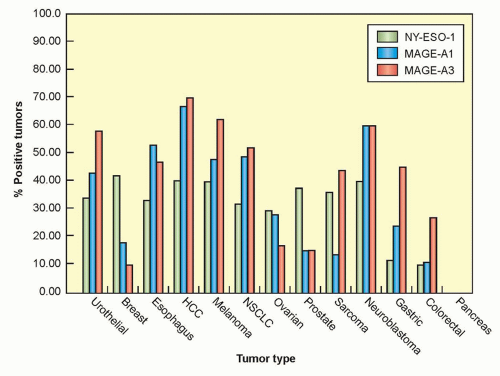 Figure 14.2 Expression of three different cancer/testes antigens in many different tumor types is shown. These data reflect reverse transcription-polymerase chain reaction measurements and is more sensitive than results obtained by immunohistochemistry. NSCLC, non-small-cell lung cancer. (Data compiled by Dr. J. Wargo. Massachusetts General Hospital.) |
Melanocyte Differentiation Antigens
Melanoma-reactive T cells have been frequently found to recognize gene products, termed melanocyte differentiation antigens (MDA), that are expressed in melanomas as well as in normal melanocytes present in the skin, eye, and ear but not in other normal tissues or tumor types. These include epitopes derived from gp100,14,15 tyrosinase,16 TRP-1,17 and TRP-2,18 proteins that had previously been found to play important roles in melanin synthesis. The screening of melanoma cDNA libraries with an HLA-A2-restricted tumor reactive T cells lead to the isolation of a previously unidentified gene, termed MART-119 or Melan-A.20 The MART-1 antigen, which is expressed in 80% to 90% of fresh melanomas and cultured melanoma cell lines as well as normal melanocytes, represents an MDA of unknown function. The majority of melanoma reactive, HLA-A2-restricted tumor-infiltrating lymphocytes (TIL) recognize a single MART-1 epitope.21 Studies carried out using a variety of approaches have also resulted in the identification of human leukocyte antigen (HLA) class II restricted epitopes of tyrosinase, TRP-1, TRP-2, and gp100.9
Overexpressed Gene Products
Gene products that are expressed at low levels in a variety of normal tissues but are overexpressed in a variety of tumor types have also been shown to be recognized by T cells. Screening of an autologous renal carcinoma cDNA library with a tumor reactive, HLA-A3-restricted T-cell clone resulted in the isolation of FGF5,22 a protein that was expressed only at low levels in normal tissues but upregulated in multiple renal carcinomas as well as prostate and breast carcinomas. The peptide epitope recognized by FGF5-reactive T cells was generated by protein splicing, a process in which distant protein regions are joined together in the proteasome that had previously only been described in plants23 and unicellular organisms.24 Subsequent studies have led to the identification of multiple epitopes that result from protein splicing, suggesting that this represents a general mechanism for generating T-cell epitopes.25,26,27,28 Screening of an autologous cDNA library led to the identification of a previously unknown gene that was termed PRAME.29 This gene product was expressed in relatively high levels in melanomas as well as in additional tumor types but was also expressed at lower levels in a variety of normal tissues that included the testis, endometrium, ovary, and adrenals. The HLA-A24-restricted PRAME reactive T-cell clone, however, expressed the natural killer (NK) inhibitory receptor p58.2, and tumor cell recognition was dependent on the loss of expression of the HLA C*07 allele that represented the ligand for the inhibitory receptor, which may explain the lack of recognition of normal tissues that express relatively high levels of this HLA gene product.
Attempts have also been made to generate T cells directed against overexpressed candidate antigens by repeatedly stimulating PBMC in vitro with peptides that were identified as high binders for particular MHC molecules either using direct binding assays or in silico analysis carried out using peptide/MHC binding algorithms.30,31 Using this approach, candidate epitopes have been identified from a variety of proteins that include prostatespecific antigen (PSA)32 and prostate-specific membrane antigen (PSMA),33 as well as Her-2/neu, a protein that is frequently overexpressed in a variety of tumor types, including breast carcinomas. Initial studies indicated that T cells derived by in vitro stimulation with a peptide that was predicted to bind with high affinity to HLA-A* 02:01, Her-2/neu:369-377, recognized the appropriate natural tumor targets.34 In one study, T cells generated following two in vitro stimulations of postvaccination PBMC from three of the four patients who were tested efficiently recognized peptide-pulsed targets but failed to recognize appropriate tumor targets.35 Similarly, although stimulation with a peptide corresponding to amino acids 540 through 548 of the human telomerase reverse transcriptase (hTERT) catalytic subunit was initially reported to generate tumor-reactive T cells,36 additional observations indicated that T cells generated using this peptide failed to recognize tumor targets.37 These factors responsible for these discrepancies remain unresolved, although the in vitro stimulation of T cells with target cells pulsed with relatively high peptide concentrations could have led to the generation of low-avidity T cells that were incapable of recognizing naturally processed antigens.
Alternative screening approaches employed for tumor antigen discovery that may help to address these issues include the use of tandem mass spectrometry to sequence peptides that have been eluted from tumor cell surface MHC molecules. Use of this technique, coupled with microarray gene expression profiling, resulted in the identification of peptides derived from proteins that appeared to be overexpressed in tumor cells.38 Peptides identified using this approach may, in many cases, not be immunogenic due to the fact that their expression in normal tissues, although lower than in tumor cells, may be high enough to lead to central or peripheral tolerance. Nevertheless, one of the peptides that were identified in this study also appeared to be recognized by human tumor reactive T cells. Recently, a similar approach was used to identify candidate peptides presented on cell surface MHC molecules that appeared to be derived from proteins that were overexpressed on glioblastomas.39 In a clinical trial involving vaccination of patients with pools of the identified peptides, overall survival was associated with the number of peptides in the vaccine pool that elicited immune response40; however, this may simply reflect the fact that T cells from healthier patients can more readily generate peptide-specific responses.
Transgenic mice that express human HLA molecules have also been immunized with candidate antigens in an attempt to identify high avidity tumor-reactive T cells. Immunization of transgenic mice expressing HLA-A*0201 with the native human p53:264-272 peptide that differed from the corresponding murine p53 sequence at a single position lead to the generation of T cells that recognized tumor cells expressing high levels of p53.41 Human T cells transduced with a murine p53 TCR isolated from an immunized mouse recognized a variety of human tumor cells; however, transduced T cells also recognized normal cells expressing lower p53 levels, indicating the dangers of targeting a normal self-protein whose expression is not strictly limited to tumor cells.42 Similarly, a TCR that was highly reactive with HLA-A*02:01+ tumor cells expressing the human carcinoembryonic antigen (CEA), a protein that is overexpressed in colon and breast carcinomas, was isolated by immunizing HLA-A*02:01+ transgenic mice with the CEA:691-699 peptide.43 The adoptive transfer of human PBMC transduced with the CEA-reactive TCR lead to an objective clinical response in one of the three treated patients; however, severe colitis was observed in all three of the treated patients.44 In general, immunotherapies that target antigens present even in small amounts on normal tissues have led to normal tissue destruction and must be applied with caution.
Mutated Gene Products Recognized by CD8+ and CD4+ T Cells
A variety of mutated antigens have also been identified as targets of tumor reactive T cells. The majority of mutated antigens identified using these approaches appear to be unique or only expressed in a relatively small percentage of cancers, and so do not
represent targets that are broadly applicable to the treatment of multiple patients. Nevertheless, these studies have in some cases provided insights into mechanisms involved with tumor development, as the mutations may represent drivers of the transformed phenotype. The CDK4 gene product that was cloned using a CTL clone contained a point mutation that enhanced the binding to the HLA-A2 restriction element.45 This mutation, which was identified in 1 of an additional 28 melanomas that were analyzed, led to the inhibition of binding to the cell cycle inhibitory protein p16INK4a and may have played a role in the loss of growth control in this tumor cell. A point-mutated product of the β-catenin gene, containing a substitution of phenylalanine for serine at position 37, was isolated by screening a cDNA library with an HLA-24-restricted, melanoma reactive TIL.46 This mutation was found to stabilize the β-catenin gene product by altering a critical serine phosphorylation site, and 2 of 24 additional melanoma cell lines were found to express transcripts with identical mutations.47
represent targets that are broadly applicable to the treatment of multiple patients. Nevertheless, these studies have in some cases provided insights into mechanisms involved with tumor development, as the mutations may represent drivers of the transformed phenotype. The CDK4 gene product that was cloned using a CTL clone contained a point mutation that enhanced the binding to the HLA-A2 restriction element.45 This mutation, which was identified in 1 of an additional 28 melanomas that were analyzed, led to the inhibition of binding to the cell cycle inhibitory protein p16INK4a and may have played a role in the loss of growth control in this tumor cell. A point-mutated product of the β-catenin gene, containing a substitution of phenylalanine for serine at position 37, was isolated by screening a cDNA library with an HLA-24-restricted, melanoma reactive TIL.46 This mutation was found to stabilize the β-catenin gene product by altering a critical serine phosphorylation site, and 2 of 24 additional melanoma cell lines were found to express transcripts with identical mutations.47
The observation that immunization against individual murine tumors did not generally cross-protect against challenge with additional syngeneic murine tumors has provided support for the hypothesis that mutant T-cell epitopes represent the predominant antigens responsible for tumor rejection.48 Mutated epitopes also represent a foreign antigen, which may render them more immunogenic than the majority of normal self-antigens. Although many of the mutations are specific for individual tumors, T cells have been generated by carrying out in vitro sensitization with peptides encoded at mutational hot spots present in driver genes.49
Recently, novel approaches have been developed that involve the sequencing of tumor cell DNA to identify potential mutated epitopes. In one study, whole exome sequencing of the murine B16 melanoma led to the identification of mutated epitopes that elicited a T cell that appeared to specifically recognize the mutated but not the corresponding wild-type peptides.50 In a second study, a mutated antigen was identified by screening candidate epitopes that were expressed by tumors derived from immunodeficient mice that regressed in immune-competent mice.51 More recently, melanomas from three patients who responded to adoptive immunotherapy were subjected to whole exome sequencing, followed by in silico analysis using peptide/MHC binding algorithms to identify candidate epitopes that were predicted to bind to the patients’ MHC molecules.52 Using this approach, a total of seven peptides were identified as targets of the TIL that were administered to these patients. Two mutated epitopes were recently identified by whole exome sequencing of a melanoma from a patient who demonstrated a partial response to treatment with the anti-CTLA-4 antibody ipilimumab, followed by a screening of a panel of mutated candidate peptide/MHC tetramers that were predicted to bind to the patient’s HLA-A and B alleles.53 In addition, a mutated epitope expressed by a bile duct cancer was identified by screening tandem minigenes encoding all mutated epitopes that were identified by whole exome sequencing.54 The adoptive transfer of T cells directed against this mutationmediated regression of the patient’s cancer. Mutations unique to each cancer represent ideal targets for immunotherapy and can potentially lead to the development of personalized therapies directed against these unique targets.
Antigens Identified in Viral-Associated Cancers
Viruses do not appear to play a role in the development of the majority of human cancers; however, an infection with HPV, a group of double-stranded DNA viruses that infect squamous epithelium, is highly associated with the development of a variety of genital lesions that range from warts to carcinomas, as well as the majority of oropharyngeal carcinomas. Recombinant vaccines have been produced by the generation of viruslike particles (VLP), self-assembling particles that form following the expression of the HPV L1 protein in recombinant viral and yeast systems that were initially found to be protective in animal models. The results of a phase II trial in which 2,392 women between 16 and 23 years of age were immunized with HPV-16 VLPs indicated that 100% of those who were vaccinated were protected against infection with HPV-16.55,56 Although vaccination with VLP does not lead to the regression of established disease, some success has been seen in therapeutic vaccination trials that target the oncogenic viral proteins E6 and E7. In a trial involving the vaccination of women with HPV-16-positive high-grade vulvar intraepithelial neoplasia with synthetic long peptides that encompass both HLA class I and class II restricted epitopes from the oncogenic HPV proteins E6 and E, clinical responses were observed in 15 of the 19 vaccinated patients, and complete regression of all lesions were seen in 9 of the 19 patients in this trial.1
Targeting foreign antigens thus may represent a strategy that can lead to more effective immunotherapies. These include viral epitopes as well as mutated epitopes that are also foreign to the host and therefore may represent more effective targets for these therapies than normal self-antigen.
HUMAN CANCER IMMUNOTHERAPIES
A wide variety of therapies have been evaluated in model systems and are now being developed for the treatment of patients with cancer. These include nonspecific approaches, those that involve direct immunization of patients with a variety of immunogens and approaches that involve the adoptive transfer of activated effector cells (Table 14.1). Much confusion related to the effectiveness of cancer immunotherapy has resulted from the lack of proper evaluation of the results of therapy using standard, accepted oncologic criteria such as the World Health Organization or the Response Evaluation Criteria in Solid Tumors (RECIST). Many clinical trials reported a positive use of soft criteria such as lymphoid infiltration or tumor necrosis that can occur in the natural course of cancer growth. Because of the delayed responses seen with some immunotherapy approaches, including tumor regression after initial tumor growth, guidelines have been published suggesting the use of an alternate set of immune-related response criteria for the evaluation of immune-based cancer treatments.57,58 Other confusion has arisen from the use of inappropriate animal models. Although animal model systems have provided important clues that may lead to improved therapies, model systems that employ artificially introduced foreign antigens or that evaluate protection from tumor challenge do not appear to be relevant to the treatment of patients with bulky metastases. Short-term lung metastasis models involve the treatment of relatively small, nonvascularized tumors and also may not be directly relevant to the majority of tumors that are the targets of current clinical trials.
TABLE 14.1 Three Main Approaches to Cancer Immunotherapy | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Nonspecific Approaches to Cancer Immunotherapy
Progress has surged in the past 10 years in the understanding and utilization of nonspecific immune stimulation for the treatment of metastatic cancers. These agents aim to activate quiescent tumor-reactive immune cells or to remove inhibitory mechanisms to allow immunosuppressed cells to function to their full capacity. Although IL-2 and ipilimumab are currently the only immune stimulants approved by the U.S. Food and Drug Administration (FDA) for the treatment of metastatic renal cell carcinoma (IL-2) and melanoma (IL-2 and ipilimumab), new immune checkpoint inhibitors such as anti-programmed cell death 1 (anti-PD-1) have shown impressive results in recent clinical trials for patients with melanoma, renal cell cancer, and also non-small-cell lung cancer (NSCLC), and will likely be approved in the near future. As expected with nonspecific immunostimulation, systemic and bystander immunerelated adverse events such as colitis has been reported with all agents in varying degrees, although most side effects are controllable and reversible if addressed aggressively and promptly by experienced clinicians. Importantly, antitumor responses seen with these immune-based modalities appear to be durable for some patients and may even be potentially curative. As with many therapies for metastatic solid tumors, preliminary trials using combination therapies have suggested better than expected response rates and survival, and confirmatory trials are in process to validate and ensure that toxicities from combining agents would not be prohibitive. Overall, patients with metastatic solid tumors may soon have wider armamentarium of off-the-shelf immunotherapy options.
Interleukin-2
Morgan et al.59 showed that a factor produced in the medium from stimulated normal human blood lymphocytes can allow ex vivo growth and expansion of human T lymphocytes. The identification of this soluble T-cell growth factor (IL-2)60,61 allowed the ability to culture T cells in vitro. IL-2 is a 15-kd glycoprotein produced in minute amounts by activated peripheral blood lymphocytes, and even with using T-cell hybridomas, minimal quantities could be purified; thus, research using IL-2 was impeded by the limited amounts of purified IL-2 available. The isolation of the cDNA clone in 198362 enabled the development in 1984 of recombinant IL-2,63 which permitted the ability to mass manufacture IL-2. Although murine studies demonstrated the ability of IL-2 to mediate tumor regression,64 early phase I clinical trials did not show any antitumor response,65 but was instructive in showing pharmacokinetics and toxicities, which led to more effective regimens. Subsequently, IL-2 was given in higher doses (up to 720,000 IU/kg intravenously every 8 hours) in a landmark trial involving 25 patients, along with nonspecific lymphokine-activated natural killer (LAK) cells, which are non-T and non-B lymphocytes.66 This report was the first to document the regression of advanced solid cancers (melanoma, renal cell, lung, and colon) using immunotherapy in humans.66 A follow-up trial randomizing 181 patients to either high-dose IL-2 alone (720,000 IU/kg intravenously every 8 hours) or high-dose IL-2 and LAK cells showed that the tumor response was due to IL-2 alone and not to the nonspecific LAK cells.67 This study also narrowed the IL-2-sensitive histologies to melanoma and renal cell cancer, which had more consistent responses.
IL-2 Therapy for Metastatic Renal Cell Cancer
Subsequent to the studies discussed previously, high-dose IL-2 was tested by additional centers and in combination with other agents for renal cell cancer. A randomized phase II trial involving 99 kidney cancer patients showed no increase in antitumor responses with the addition of interferon alfa-2b (IFNα-2b). Responses were seen for 12 (17%) of 71 patients who received high-dose IL-2 alone, with 4 complete regressions.68 A summary report of 227 patients with metastatic renal cell cancer treated with high-dose IL-2 (defined as 600,000 IU/kg or 720,000 IU/kg given intravenously every 8 hours as tolerated up to 15 doses) from 1985 to 1996 at the Surgery Branch of the National Cancer Institute (NCI) documented a total response rate of 19%, with 10% partial and 9% complete; the longest duration of a complete response was over 10 years ongoing (134+ months).69 Another summary report from seven phase II clinical trials from multiple institutions involving 255 patients with metastatic renal cell cancer receiving high-dose IL-2 showed the overall response rate was 14%, with 9% partial and 5% complete, and responses occurred in all sites of disease, including primary kidney tumors, bone metastases, and bulking visceral tumor burdens.70 Although the response rates were modest, the durability of the responses was remarkable, with many responses lasting over 5 years ongoing (see Fig. 14.2). Because of the striking durability of the antitumor responses, IL-2 received FDA approval for the treatment of metastatic renal cell cancer in 1992. A follow-up report in 2000 showing the response rates of the 255 renal cell patients in the seven phase II studies to be the same, with complete responses lasting over 10 years ongoing (131+ months for the longest responder), suggesting a potential cure.71
To ascertain whether lower doses and/or different administration routes, which would decrease toxicity and obviate the need for inpatient hospitalization for IL-2 therapy, a trial randomizing 400 patients with metastatic renal cell cancer to either standard high-dose intravenous IL-2, low-dose intravenous IL-2 (at 72,000 IU/kg), or low-dose subcutaneous IL-2 (250,000 U/kg per dose daily Monday through Friday in the first week and then 125,000 U/kg per dose daily during the next 5 weeks).72 Although responses were seen with all three regimens, including complete responses in the low-dose subcutaneous regimen, standard highdose IL-2 had higher overall response rates (21%) versus low-dose intravenous IL-2 (13%; p = 0.048) and low-dose subcutaneous IL-2 (10%; p = 0.033), suggesting the superiority of the high-dose intravenous regimen.72
The administration of IL-2 represents the only known curative treatment for patients with metastatic renal cell cancer and should be considered as front-line therapy for suitable patients.
IL-2 Therapy for Metastatic Melanoma
Between 1985 and 1993, 270 patients with metastatic melanoma enrolled into eight clinical trials in multiple centers using highdose IL-2 (defined as 600,000 IU/kg or 720,000 IU/kg given intravenously every 8 hours as tolerated up to 15 doses). Atkins et al.73 reported overall response rates of 16% (43 patients), with 10% partial and 6% complete; responses occurred at all tumor sites and regardless of initial tumor burden. With median follow-up at that time of 62 months, 20 responders (47%) were still alive, with 15 surviving over 5 years.73 A follow-up report on those patients in 2000 showed that the response rates were unchanged; with the longest response duration of >12 years ongoing, disease progression was not observed in any patient responding greater than 30 months.74 As with renal cell cancer, the flat tail of the Kaplan-Meier response duration and overall survival curves (Fig. 14.3), showing the potential curative nature of the antitumor responses, was the main compelling reason the FDA approved IL-2 for the treatment of metastatic melanoma in 1998.
Research in subsequent years aimed to increase the response rates of IL-2, led by increasing interests in tumor vaccinations as melanoma-associated antigens were being characterized.75 Pilot studies suggested that vaccinations using modified melanoma differentiation antigens such as gp100:209-217(210M) could elicit immunologic responses in nearly all patients, and when combined with high-dose IL-2, could elicit potentially higher than expected clinical antitumor responses.75 A follow-up phase III study76 randomized 185 patients with HLA*A0201 from 21 centers to either high-dose IL-2 or high-dose IL-2 plus gp100:209-217(210M) concurrent immunization. Although the response rates for the
IL-2 plus vaccine arm was statistically improved compared to IL-2 alone (16% versus 6%; p = 0.03), the IL-2 alone arm was notable for being much lower than in all prior studies.76 In addition, a pilot trial of 36 melanoma patients treated high-dose IL-2 concurrently with ipilimumab (an antibody against cytotoxic T lymphocyte-associated antigen 4 discussed in the following section) gave a 25% OR rate, with 17% achieving complete response77; however, these data have not been further tested.
IL-2 plus vaccine arm was statistically improved compared to IL-2 alone (16% versus 6%; p = 0.03), the IL-2 alone arm was notable for being much lower than in all prior studies.76 In addition, a pilot trial of 36 melanoma patients treated high-dose IL-2 concurrently with ipilimumab (an antibody against cytotoxic T lymphocyte-associated antigen 4 discussed in the following section) gave a 25% OR rate, with 17% achieving complete response77; however, these data have not been further tested.
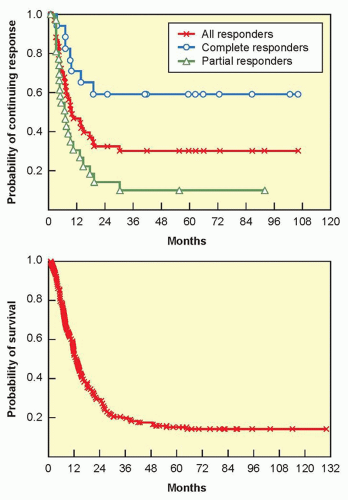 Figure 14.3 Kaplain-Meier plots of response duration (top) and overall survival (bottom) for 270 patients with metastatic melanoma who were treated with high-dose bolus IL-2 from 1985 to 1993 in eight clinical trials.73 |
Correlative studies suggest that the total doses of IL-2 received during the first treatment course was significantly higher in patients achieving a complete response69; however, when limited to patients who were able to complete both cycles of the course, there was no statistical significance, suggesting that patients whose tumors progressed significantly after one cycle (and was not able to complete the second cycle of the course) accounted for some of the difference seen.78 Responders did have a higher maximal lymphocyte count69,78 immediately posttherapy and were more likely to develop vitiligo and thyroid dysfunction.78 There has not been a consistent pretherapy factor that is predictive of response, although one retrospective correlative study involving 374 patients showed that patients with M1a (subcutaneous- and/or cutaneous-only disease) have a response rate of 54% compared with 12% for those with visceral M1b/c (P2 <0.0001).78
Toxicities and Safe Administration of IL-2
High-dose IL-2 has been shown to be associated with adverse events that impact multiple organ systems.73,79,80 The main component of the toxicities is due to an inflammatory response mediated by the release of cytokines such as IFNγ and tumor necrosis factor alpha (TNF-α)81 resulting in a capillary-leak syndrome82 and decreased systemic vascular resistance, which can lead to fever, hypotension, cardiac arrhythmia, lethargy, renal insufficiency, hepatic dysfunction, body edema, pulmonary edema, and confusion; other side effects can also include nausea, diarrhea, rash, anemia, thrombocytopenia, lymphocytosis, and neutrophil chemotactic defect83 that predispose patients to gram-positive line infections. Since the first clinical trials with IL-2 in 1984, however, much has been learned to permit its safe dosing for appropriately screened patients82,84,85; importantly, if patients are appropriately supported, side effects are quickly reversible once IL-2 dosing ceases.85 Kammula et al.86 compared the incidences of grade 3/4 toxicities between the 155 patients treated from 1985 to 1986 to 156 patients treated from 1993 through 1997 at the NCI Surgery Branch: grade 3/4 hypotension decreased from 81% to 31%, intubations from 12% to 3%, neuropsychiatric toxicities from 19% to 8%, diarrhea from 92% to 12%, line sepsis from 18% to 4%, cardiac ischemia from 3% to 0%, and mortality from 3% to 0%. In fact, no fatality occurred strictly due to IL-2 therapy since 1989.86 Overall strategies for the safe administration of high-dose IL-2 include careful screening for appropriately selected patients with adequate cardiopulmonary reserve, having an experienced team of physicians and nurses who are cognizant of the expected toxicities of IL-2, having routine preemptive measures such as prophylactic antibiotics to prevent line infections, and aggressive and prompt management of toxicities.
Checkpoint Modulators
Anti-Cytotoxic T Lymphocyte Antigen 4
CTLA-4 is an immunosuppressive costimulatory receptor found on newly activated T cells (and on regulatory T cells) that binds with costimulatory ligands B7-1 and B7-2 on antigen-presenting cells.87,88 When CTLA-4 is engaged by B7-1 or B7-2, the T cells becomes inhibited,89,90 suggesting that CTLA-4 likely evolved as a self-protective mechanism to prevent autoimmunity (Fig. 14.4). Thus, overcoming this checkpoint molecule was an aim of cancer immunotherapy. After CTLA-4 blockade in murine models led to antitumor immunity,91,92 anti-CTLA-4 antibodies were tested in clinical trials starting in 2002.
The combination of anti-CTLA-4 blocking antibodies and vaccination worked well in murine models and led to one of the early phase II studies using ipilimumab (a fully human immunoglobulin [IgG1] monoclonal antibody previously called MDX-010) with two gp100 vaccines, gp100:209-217(210M) and gp100:280-288(288V), in patients with metastatic melanoma.93 Antitumor regressions were seen (from 11% to 22% overall response rates, with up to 8% complete response rates), along with severe autoimmune toxicities such as colitis, dermatitis, and even hypophysitis,93,94,95 as would be expected based on the mechanism of CTLA-4 blockade. In fact, autoimmunity adverse events appeared to correlate with response to ipilimumab.3 The experience with these early studies led to management strategies to screen aggressively for immune-related adverse events (IRAE), such as routine screening of endocrinopathies, and to treat IRAEs promptly, including high-dose steroids if needed for severe colitis.96,97 Overall, ipilimumab was in some ways easier to manage for the patients than IL-2 because it was an outpatient infusion given every 3 weeks; IRAEs were unpredictable, however, and can appear suddenly many weeks after receiving a dose.
In 2010, results from a landmark phase III randomized trial comparing three treatment strategies (ipilimumab alone, gp100 peptide vaccine alone, or ipilimumab plus gp100 peptide vaccine) in 676 patients with metastatic melanoma were published showing improvement in median survival in the two arms that received ipilimumab (10 months) compared to the gp100 alone arm (6 months, p <0.001), despite showing a low response rate of 7% (among 540 patients who received ipilimumab).98 Another
phase III randomized trial comparing dacarbazine plus ipilimumab versus dacarbazine alone again showed improved survival in that arm containing dacarbazine (11.2 months versus 9.1 months; p <0.001).99 These studies showing survival benefit led to FDA approval of ipilimumab for advanced melanoma in 2011.
phase III randomized trial comparing dacarbazine plus ipilimumab versus dacarbazine alone again showed improved survival in that arm containing dacarbazine (11.2 months versus 9.1 months; p <0.001).99 These studies showing survival benefit led to FDA approval of ipilimumab for advanced melanoma in 2011.
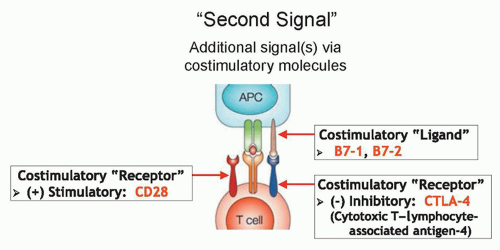 Figure 14.4 Mechanism of action of cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4). When CD28 is engaged on the T cell, reactivity of the T cell is enhanced. When CTLA-4 is engaged on the T cell, reactivity of the T cell is inhibited. Blocking of CTLA-4 with a monoclonal antibody can elicit antitumor immunity but also autoimmunity. |
The responses seen with ipilimumab appear to be durable.100 A follow-up study of 177 patients with metastatic melanoma treated on the earliest trials at the NCI Surgery Branch using ipilimumab showed that response duration could last 99+ months ongoing.77 In fact, 14 out of the 15 complete responders remain disease free 54+ to 99+ months ongoing, suggesting a potential cure for some patients. Interestingly, several patients who were deemed partial responders converted to complete responders several years later, because it look an average of 30 months to have all visible tumor marks on imaging scans to disappear.77
Ipilimumab was also tested on other solid tumors, and renal cell cancer again appears to be the only other type beside melanoma that had significant responses. Sixty-one patients with metastatic renal cell cancer were treated, and six developed a response (10%); however, 33% developed grade 3/4 IRAEs.101 Subsequently, the availability of agents with lower toxicity profiles such as sunitinib and sorafenib prevented further enthusiasm to pursue this drug for renal cell cancer.
Another anti-CTLA-4 antibody, tremelimumab (previously called CP-675,206), has also demonstrated durable responses in melanoma patients.102,103 A phase III randomized trial randomizing 655 patients with metastatic melanoma to either tremelimumab or physician’s choice chemotherapy, however, failed to show a survival difference (despite a significantly different response duration favoring tremelimumab, 35.8 months versus 13.7 months; p = 0.0011), possibly due to crossover of chemotherapy patients enrolling into ipilimumab trials and expanded access programs.103
Anti-Programmed Death 1 and Anti-Programmed Death Ligand 1
PD-1 is another checkpoint modulator expressed on activated T cells. Although CTLA-4 appears to be involved in the early activation of T cells, PD-1 is involved in the later effector phase of T-cell activation and can function to prevent excessive damage to self by activated T cells in the periphery.104,105 Interaction with its corresponding ligand, PD-L1 (B7-H1) and PD-L2 (B7-H2) leads to suppressed T-effector function. PD-L1 is expressed on hematopoietic and epithelial cells and is upregulated by cytokines such as IFNγ,106 whereas PD-L1 is mainly on antigen-presenting cells. Given the clinical results with inhibiting the CTLA-4 checkpoint, recent efforts have focused on inhibiting the PD-1/PD-L1 and PD-1/PD-L2 interactions.
Nivolumab (previously known as BMS-936558, MDX-1106, and ONO-4538) is a fully human anti-PD-1 IgG4 monoclonal antibody that was initially tested in a phase I trial published in 2010 in which 39 patients with advanced solid cancers were treated in escalating doses.107 Responses were seen in one patient with colon cancer, one with melanoma, and one with renal cell cancer; one patient developed colitis.108 These hopeful results lead to a larger study in which 236 patients with either NSCLC (74 patients), melanoma (94 patients), or renal cell cancer (33 patients).4 Objective responses were seen in 18% of patients with NSCLC, 28% with melanoma, and 27% with renal cell cancer.4 Grade 3/4 adverse events occurred in 14% of patients, including those previously seen with ipilimumab (dermatitis, colitis, hepatitis, thyroiditis, hypophysitis, and pneumonitis). Nine patients developed pneumonitis, six of whom was reversible, and three (1%) with grade 3/4 died despite steroids and infliximab therapy.4 An update on the status of 107 melanoma patients treated from 2008 to 2012 shows a 31% tumor response rate, with a median response duration of 2 years and a median overall survival of 16.8 months.109
Nivolumab was also tested in combination with ipilimumab in melanoma in either concurrent (53 patients) or sequenced (33 patients) regimens. The concurrent group experienced an overall response rate of 40%, whereas the sequenced group had a 20% response rate.110 The concurrent group also experienced a higher rate of grade 3/4 adverse events (53%), compared to 18% in the sequenced group. Interestingly, 16 of 21 responders in the concurrent group experienced tumor reduction of 80% or greater by 12 weeks,110 a tempo that is faster than was seen with ipilimumab.
Another anti-PD-1 developed independently, lambrolizumab (previously known as MK-3475, a humanized IgG4κ monoclonal antibody), was tested on 135 patients with metastatic melanoma.111 The response rate was found to be 38% and was similar between those who had received ipilimumab and those who were ipilimumab naïve,111 confirming that the antitumor response from lambrolizumab occurs via a different mechanism. Similar to nivolumab, 13% of patients developed grade 3/4 adverse events, with 4% developing pneumonitis, although none developed grade 3/4 pneumonitis.111
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


