Conrad Hal Waddington is usually credited with modifying the Greek word “epigenesis,” which described a theory of development, into a new term “epigenetics” to mean “the causal interactions between genes and their products which bring the phenotype into being.”1 Waddington described an “epigenetic landscape” usually as a system of bifurcating valleys through which a cell, depicted alternatively as water or a roulette ball, flows or rolls toward the sea constantly making binary choices on its way.2 Waddington explained how genes were responsible for creating this “epigenetic landscape” of valleys since these genes were positioned on the underside of the landscape and attached to it by a series of “guy ropes” much like how pegs with ropes determine the shape of a tent’s canopy. Thus, genes were ultimately responsible for the motley array of crevices, valleys, and hills in the canopy through which a ball could follow downhill as it embarked on its developmental pathway of differentiation. In addition to Waddington, other leading biologists of the time, including Ernest Hadorn, Richard Goldschmidt, and Julian Huxley, also saw a relationship between genes and their action to development during a period when embryology and developmental biology were very disparate disciplines.4 Thus, the term “epigenetics” proved durable not only because it provided a convenient explanation of how expressed genes informed developmental decisions of a cell, but also because it brought together the two disciplines of Mendelian genetics and embryology. In time, epigenetics has become used not only to explain heritable changes in development, but also to understand diverse biological processes including normal aging, maternal X chromosome inactivation, as well as pathological states, such as cancer and neurodegenerative diseases.
In our modern experimental, scientific era, it has become apparent that it is not only the information contained within genes that is important (the so-called “hardware”), but also how genes are organized or packaged within the cell (the “software”) that is critical. This is because the precise, compact folding of the DNA into its chromatin structure has a significant effect on which genes are actively transcribed by DNA polymerase and which are not (largely, it can be thought of as an accessibility problem—the more compactly folded or closed the DNA, the more inaccessible to being read or transcribed). The contemporary definition of epigenetics as “a hereditable change in gene expression that occurred without a change in the DNA sequence”5 can also be attributed to work done by both Robin Holliday and Arthur Riggs in the 1970s. Both authors independently published papers suggesting that DNA methylation may be responsible for an epigenetic switch between activity and inactivity of genetic transcription that affected cell development.6,7 In 1987, Holliday revisited Waddington’s original terminology and applied it to changes in gene expression caused by DNA methylation.8 Riggs followed in 1996 stating more broadly that epigenetics was “the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence.”9
Although epigenetics is evident in all biological processes, it is in cancer, and indeed in thoracic malignancies, where some of the translational progress from the bench to the clinic has been taking place. Nowadays, epigenetics refers to both physical and chemical ways of modifying chromatin to control gene expression. In contemporary cancer biology, epigenetics has been expanded to include at least four main, regulatory mechanisms that affect gene expression via DNA transcription: (1) DNA methylation, (2) chromatin remodeling via posttranslational histone covalent modifications, (3) chromatin modifications via noncovalent modifications such as nucleosome repositioning and histone variants, and (4) deregulation of MicroRNA Activity.10 DNA methylation is the most widely known epigenetic alteration having been first discovered in the 1960s by Griffith and Mahler.4 Simply stated, (1) most cancers are hypomethylated if all the DNA is considered, (2) hypermethylation (or just methylation) of a gene’s promoter region occurs in mainly in malignancy and rarely in nonmalignant DNA, and (3) transcription silencing and no protein expression is the result of DNA promoter methylation.
This refers to the postsynthetic covalent addition of a methyl group to cytosine mediated by three known active DNA methyltransferases (DNMTs). It plays a critical and essential role during normal embryogenesis enabling genomic imprinting, X-chromosome inactivation, and suppression of transposons, as well as repetitive elements. It has been implicated in a wide variety of human biological processes including aging, atherosclerosis, and neurobiology, and is considered to be a probable mediator between nature and nurture. But it is the role of DNA methylation in carcinogenesis which we will briefly focus on here. Noncancerous and cancerous DNA differ both in the overall number of methyl groups attached to their DNA as well as by the frequency of methylation in specific areas, especially the promoter region of certain genes. The first change in methylation noted as early as 1983 was that most cancers tended to have fewer methyl groups attached to their DNA11,12 than noncancerous DNA. But the most well studied difference between cancer and noncancer DNA has been that hypermethylation is observed in cancers at the promoter region of certain genes.13 This hypermethylation tends to occur at CpG dinucleotides in the promoter in areas known as CpG islands which are usually between 500 bp and 2000 bp in length. What has been widely documented is that there is a reciprocal relationship between the density of methylated cytosine residues in the gene promoter and the transcriptional activity of that gene. In general, methylated genes prevent the transcriptional machinery, including DNA polymerase, from accessing the promoter to transcribe the DNA into mRNA, and the gene becomes transcriptionally silent. Moreover, it is probable that the density of methylation increases the longer and more permanent the gene remains silenced. Unmethylated DNA, however, is open, accessible, and transcriptionally active.
The mammalian DNA double helix is wound around a scaffold of histone proteins (like thread wound around spools) and the tightness of the wrap depends on chemical projections that extend from the side of the spools, the so-called histone tails (Fig. 7-1). Certain chemical modifications of these histone tails cause the DNA to wound even more tightly. The tighter the wrap, the more inaccessible the DNA is to being read or transcribed by DNA polymerase (Fig. 7-1A). So, like DNA methylation, histone modifications represent the epigenetic regulation of DNA transcription. The result here is that a gene encoded by a tightly wound DNA is not expressed. The best characterized histone modifications are acetylation and methylation. In due course, there was a recognition that there was a certain pattern of histone modifications or a histone codes (often the acetylation or methylation of certain histone lysine residues) that were responsible for determining which genes of the DNA sequence are active or silent. Acetylation of histones is usually associated with euchromatin because it weakens the attraction of the positively charged histone for the negatively charged DNA resulting in a looser, more relaxed, and accessible chromatin (Fig. 7-1B). Methylation of the histone lysines, however, can be active or repressive depending on the position of the lysine on the histone tail and the degree of methylation. H3K4 trimethylation, for example, is associated with euchromatin, whereas H3K9 or H3K27 trimethylation is usually present at the promoter of a transcriptionally repressed gene. It soon became apparent that this histone code, like DNA methylation, can also be inherited from generation to generation. Finally, there has recently been a further recognition that there are complex interactions between DNA methylation and the histone code that further complicate epigenetic regulation of transcription, and thus, gene expression.10 Although the exact mechanisms underlying these chromatin modifications are well beyond the explanations that can be provided in this chapter, enzymes responsible for H3K27 trimethylation, for example, seem to be upregulated in thoracic oncologic tumors, and inhibitors to these enzymes may have therapeutic benefit.
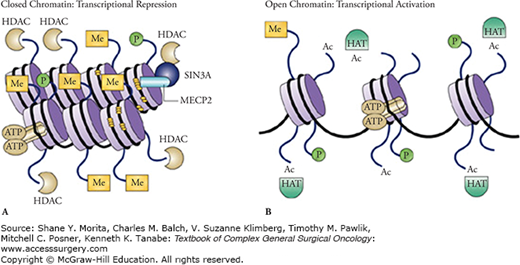
A nucleosome is composed of a segment of DNA wrapped around eight histone protein cores and represents a basic unit of DNA packaging within a cell. Nucleosomes do not randomly occur throughout the genome but are precisely organized according to transcriptional activity.14 Nucleosome-free regions are apparent just upstream from a transcription start site, whereas nucleosome-rich areas occur in promoters with gene repression.15,16 Of course, this physical nucleosomal repositioning cooperates with DNA methylation and histone modifications to promote gene activation or repression accordingly.10
Not only are histones posttranslationally modified to change chromatin, but the actual histones themselves can be displaced by the site-specific deposition of certain histone protein variants. The histone protein variant H3.3, for example, can replace existing histones and act as an upstream signal itself to change a chromatin state to one of a more active gene.17 The variant H2AZ has even been shown to promote euchromatin by protecting against DNA methylation.18
These are short, single-stranded noncoding RNA approximately 22 nucleotides in length that regulate gene expression in as many as 30% of mammalian genes. Since the first miRNA, lin-4, was observed in 1993, a flurry of new miRNA along with their target genes have been increasingly identified.19 miRNAs are incorporated into a ribonuclear particle to form an RNA-induced silencing complex which targets, in a sequence-specific manner, messenger RNA for degradation. This effectively silences a gene. Interestingly, expression of a miRNA itself can be regulated by epigenetics,10 and the interaction between miRNAs, DNA methylation, and other parts of the epigenetic machinery is increasingly being observed.
We have only briefly described each of these four mechanisms in this chapter, and their relevance to clinicians. It is suffice to note here that most of the work over the last four decades has focused on DNA methylation, investigations inspired by Holliday and Pugh’s publication in 1975.7 DNA methylation is thus the most widely known epigenetic change, especially the reciprocal relationship between the increased density of methylated cytosine residues and decreased transcriptional activity of the gene. For surgeons, it is also important to note that it has been in the understanding of DNA methylation where the large growth of translational application of epigenetic scientific principles in cancer has occurred. To date, this has extended to commercial availability of cancer biomarkers for early detection, prognosis, prediction, and even the production of epigenetic drugs for therapy.
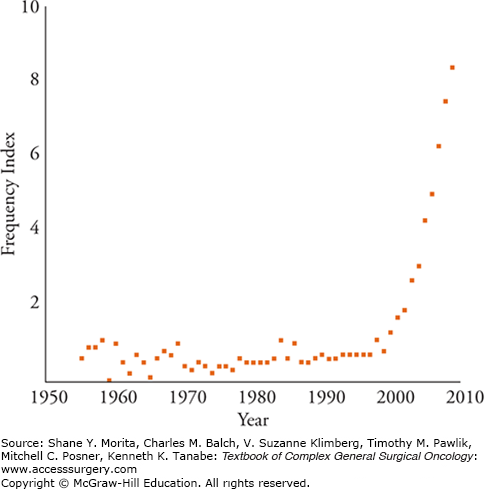
From Waddington’s coinage of the term until the late 1990s, articles using the word “epigenetics” appeared in the literature fewer than 100 times.20 Figure 7-2 shows the exponential growth of the epigenetic literature and the tenfold increase in these articles through 2009. One of the reasons for epigenetics’ sudden fame was a newfound appreciation of the complexity and importance of how our genes are expressed from DNA. Before the Human Genome Project it was surmised that given the complexity of humans, our genome must have at least 100,000 genes to account for the body’s myriad functions. At the end of the project, it was shocking to realize that not only were humans composed of only approximately 30,000 genes, but also that we were only two to three times as complex as simple laboratory organisms such as Caenorhabditis elegans and Drosophila. In fact, more recent estimates have even revised the human genome to approximately 21,000 genes.21 The main explanation that seems to account for human complexity from such a small genetic base is that the human genome has many more options for gene expression than other organisms. There is a realization that the DNA sequence or “hardware” is not the limiting factor, and that gene expression is not a simple event, but that the inner workings of transcription or the “software” are equally as important. As described above, the new science of epigenetics explains many of these transcriptional events that seem to regulate gene expression.
FIGURE 7-2
Relative frequency of articles with epigenetic or epigenetics in their title. The frequency index is the number of titles retrieved from the ISI Web of Knowledge using the search term epigenetic divided by the number of titles retrieved using genetic multiplied by 100. An index of 1 means there is one “epigenetic” title for every 100 “genetic” titles. (Reproduced with permission from Haig D. Commentary: the epidemiology of epigenetics. Int J Epidemiol. February 2012;41(1):13–16.)
Although there was an appreciation in the 1990s that epigenetics could produce distinct changes in phenotype, it was widely thought that only genetic changes were heritable and that epigenetic alterations were somewhat transient and could be cleared at the level of the germline in mammals. A surge in interest in epigenetics thus ensued when it began to be understood that epigenetic changes wrought on the DNA were heritable. This was observed not only as mitotic heritability, since it could be maintained through multiple cell divisions, but also as transgenerational heritability since the same phenotype could be transmitted to mammalian offspring. This latter concept was widely accepted after a series of experiments with the Agouti (Avy) mouse model. Wild-type mice for the Agouti gene are brown and thin, whereas the mice who have constitutive, ectopic Agouti gene expression have yellow fur, adult-onset obesity, diabetes, and exhibit tumorigenesis (Fig. 7-3). In one set of maternal exposure experiments, researchers showed conclusively that the coat color variation of mice offspring directly correlated with the level of epigenetic marks on a cryptic promoter area that controls the ectopic Agouti gene expression.22 Yellow fur correlates with no methylation (increased ectopic Agouti expression) while the fur becomes increasingly brown as the cryptic promoter region becomes progressively methylated and ectopic Agouti expression decreases. The maternal exposure (given early in embryogenesis to have the maximum effect on fetal epigenome) was either maternal nutrition due to a dietary supplement with a methyl donor genisten (the major isoflavone present in soy), which caused an increase in methylation or an environmental exposure, and bisphenol A (BPA—a chemical used in the manufacture of plastics and epoxy resins commonly found in food and beverage containers, baby bottles, and dental sealants) which caused a decrease in methylation. Not only did the genisten (methyl donor) and the BPA (demethylating agent) shift the coat color distribution of the murine offspring toward brown or yellow respectively, but the phenotypic change was marked by even an alteration in the levels of DNA methylation in tissues from the three germ layers of the pup (brain, kidney, and liver), showing that the process occurred early during their embryonic development. Moreover, the pup’s coat colors persisted into adulthood showing stable inheritance of the methylation status as well as the propensity, or lack thereof, for obesity, diabetes, or tumorigenesis.

The first translational applications of epigenetics in clinical medicine used DNA methylation as a biomarker for the early detection of cancer, for measuring the sensitivity of a cancer to a particular therapy, and for determining a patient’s prognosis. One of the reasons for the relative quick translation of DNA methylation from a basic science concept to an active application in clinical medicine was the development of an inexpensive, relatively quick, PCR-based method of detecting DNA methylation. Methylation-specific PCR was developed in 1995 at Johns Hopkins University,23 was quickly licensed for commercial use, and has become the basis of several of the first FDA-approved diagnostic and predictive assays in epigenetics.
The basic science literature is replete with a large number of publications on the potential use of DNA methylation as a marker for multiple solid cancers in many bodily tissues and fluids. Although the first several commercial products using epigenetics have not been in thoracic oncology per se, recounting their development here in this chapter is informative as to how new epigenetic markers will be commercialized for thoracic oncologic tumors. The first example is for prostate cancer while the second is for colorectal carcinoma.
In the first example, the clinical need for a more sensitive and specific test has led to the development of the epigenetic biomarker test, ConfirmDX™, that may obviate repeat biopsies needed in the practice of early detection of prostate cancer. Twenty-seven percent of men with elevated PSA tests between 3.1 to 4 ng/mL are known to harbor occult prostate cancer despite histologically negative biopsy results from a 10 to 12 core biopsy.24 Most urologists agree that only 1% of the prostate is adequately interrogated by the standard core biopsy protocols. The current standard of care in early diagnosing prostate cancer has resulted in about 40% of men undergoing a repeat biopsy a second, third, and even fourth time to ensure there is no occult cancer present. In two large, multicentered, prospective clinical trials, one European and the other North American, DNA methylation of three epigenetic biomarkers outperformed histopathological criteria in assessing prostate biopsies.25,26 Specifically, the results showed that GTSP1, APC, and RASSF1A had approximately a 90% negative predictive value in selecting subjects at low risk of prostate cancer. Remarkably, it took less than two decades from noting that pi-class glutathione S-transferase gene (GSTP1) methylation was one of the most common genomic alterations in prostate cancer to the gene being the basis of the first commercialized product to make it to market to assist in the early diagnosis of prostate cancer. In 1994, DNA promoter hypermethylation was reported to silence the GTSP1 gene’s expression effectively in 20 of 20 human prostatic carcinoma tissue specimens.27 Later, GSTP1 was confirmed to be transcriptionally inactivated in over 90% of prostate cancers, but rarely methylated in benign prostatic hyperplasia or normal prostate tissues.28 GTSP1 is involved in protecting cells as a detoxifying agent from carcinogen-mediated DNA damage. The other two genes are classic tumor suppressor genes and when they are methylated and silenced, cancer can result.
The key point here is that the ConfirmDX™ assay, based on the cancer-specific methylation of these three genes, fills a critical, clinical gap in the early detection of prostate cancer that was not being met with the traditional tools of histopathology. This is most likely because these genes are able to pinpoint a molecular, epigenetic field effect that is associated with cancer at the DNA level. Of the 40% of men who now are being referred for subsequent biopsies based solely on histopathological criteria, this epigenetic assay seems to be able to pinpoint those who actually need it.
The second example of a FDA approved, early diagnostic test for cancer that has an important epigenetic component is a stool-based molecular assay for colon cancer, called Cologard™. This in vitro test is designed to indicate individuals who have either colorectal cancer or adenomatous polyps, not only by detecting in stool DNA two methylated genes, the N-Myc downstream-regulated gene 4 (NDRG4), and the bone morphogenetic protein 3 gene (BMP3), but also by finding in the stool seven Kras point mutations, as well as fecal occult hemoglobin. The marketing of this assay is the culmination of several years of bench research,29–31 and will be offered to asymptomatic individuals 50 to 80 years ago who have no elevated risk factors for colon cancer. Its current indication is not as a substitute for a diagnostic colonoscopy, but as an adjunct with current methods of testing for colon cancer in accordance with present screening guidelines.
The pivotal study for the FDA approval of the Cologard™ assay (the Multi-Target Colorectal Cancer Screening Test for the Detection of Colorectal Advanced Adenomatous Polyps and Cancer Study or “DeeP-C”) involved 10,023 subjects at 90 sites which ultimately included 65 colorectal cancer cases and 760 cases of advanced adenomatous polyps.32 The test detected 60 of the 65 colon cancer cases which were ultimately diagnosed by colonoscopy yielding a sensitivity of 92.3%. The specificity was high at 86.6%. The colon cancers detected by the assay were weighted toward stages I to III which are precisely those stages of the disease with the highest survival if detected early. In addition, the assay proved superior to immunochemical-based fecal occult blood testing (FIT) in its sensitivity to detect colorectal cancer (92.3% vs. 73.8%). FIT detected only one cancer case missed by the Cologard™ assay, whereas the latter found 13 cases not identified by the former. Moreover, the molecular assay showed a significant advantage over FIT in detecting adenomas, particularly large lesions, and colorectal cancer.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


