Bronchial and thymic carcinoids are rare. We present epidemiologic data and potential risk factors. The approach to bronchial and thymic carcinoid patients is discussed, from the initial diagnosis and evaluations to treatment. These malignancies follow staging systems of their site of origin. Because bronchial and thymic carcinoids are rare, we use many treatment strategies that have been demonstrated in gastrointestinal and pancreatic neuroendocrine tumors. The lack of information regarding efficacy in bronchial and thymic carcinoids, as well as the scarcity of therapeutic options available, demands the importance of clinical trials that include these patients.
Key points
- •
Bronchial carcinoids are well-differentiated neuroendocrine tumors that account for approximately 2% of all lung tumors.
- •
Bronchial carcinoids are usually sporadic, but may be associated with multiple endocrine neoplasia type 1 (MEN-1) syndrome, and are classified into typical and atypical carcinoids.
- •
MEN-1 syndrome is associated with up to 25% of cases of thymic carcinoids.
- •
Small biopsies and cytology cannot distinguish between atypical and typical carcinoid; surgical specimens are often required for classification.
- •
Surgery is the primary treatment modality for patients with localized disease and is the only curative option; while in Stage IV disease, there are few recommendations as to the approach to care.
Introduction
Carcinoid tumors arising in the lung and thymus are rare foregut neuroendocrine tumors (NETs) characterized by neuroendocrine morphology and differentiation and are classified using the World Health Organization (WHO) criteria, first described in 2004 and recently updated in 2015 ( Table 1 ). At one end of the spectrum are typical carcinoids (TC), which are low-grade, well-differentiated tumors that often present in early stage, rarely metastasize after surgical resection, and are generally relatively resistant to chemotherapy. At the other end of the spectrum are high-grade, poorly differentiated tumors such as small cell or large cell neuroendocrine carcinomas, which behave aggressively, often present with distant metastasis, and are sensitive to chemotherapy. Atypical carcinoids (AC) are considered to be well differentiated NETs, although they are intermediate grade and represent a more aggressive phenotype when compared with TC. In this review, the epidemiology and risk factors, as well as diagnostic and treatment paradigms, for typical and AC tumors of the lung and thymus are discussed. Owing to the rarity of these malignancies, treatment approaches have not been validated in large studies.
| Nomenclature | Grade | Histopathologic Characteristics | Differentiation |
|---|---|---|---|
| Typical carcinoid | Low | Carcinoid morphology, <2 mitosis/2 mm 2 , no necrosis, >0.5 cm diameter | Well differentiated |
| Atypical carcinoid | Intermediate | Carcinoid morphology, 2–10 mitosis/2 mm 2 , or foci of necrosis | Well differentiated |
| Large cell carcinoma | High | NE structure, >10 mitosis/2 mm 2 , necrosis (may be extensive), cytology resembling NSCLC, IHC positive for NE markers and/or NE granules by electron microscopy | Poorly differentiated |
| Small cell carcinoma | Small cell size, scant cytoplasm, nuclei with finely granular chromatin and absent or faint nucleoli, >11 mitoses/2 mm 2 , extensive necrosis |
Introduction
Carcinoid tumors arising in the lung and thymus are rare foregut neuroendocrine tumors (NETs) characterized by neuroendocrine morphology and differentiation and are classified using the World Health Organization (WHO) criteria, first described in 2004 and recently updated in 2015 ( Table 1 ). At one end of the spectrum are typical carcinoids (TC), which are low-grade, well-differentiated tumors that often present in early stage, rarely metastasize after surgical resection, and are generally relatively resistant to chemotherapy. At the other end of the spectrum are high-grade, poorly differentiated tumors such as small cell or large cell neuroendocrine carcinomas, which behave aggressively, often present with distant metastasis, and are sensitive to chemotherapy. Atypical carcinoids (AC) are considered to be well differentiated NETs, although they are intermediate grade and represent a more aggressive phenotype when compared with TC. In this review, the epidemiology and risk factors, as well as diagnostic and treatment paradigms, for typical and AC tumors of the lung and thymus are discussed. Owing to the rarity of these malignancies, treatment approaches have not been validated in large studies.
| Nomenclature | Grade | Histopathologic Characteristics | Differentiation |
|---|---|---|---|
| Typical carcinoid | Low | Carcinoid morphology, <2 mitosis/2 mm 2 , no necrosis, >0.5 cm diameter | Well differentiated |
| Atypical carcinoid | Intermediate | Carcinoid morphology, 2–10 mitosis/2 mm 2 , or foci of necrosis | Well differentiated |
| Large cell carcinoma | High | NE structure, >10 mitosis/2 mm 2 , necrosis (may be extensive), cytology resembling NSCLC, IHC positive for NE markers and/or NE granules by electron microscopy | Poorly differentiated |
| Small cell carcinoma | Small cell size, scant cytoplasm, nuclei with finely granular chromatin and absent or faint nucleoli, >11 mitoses/2 mm 2 , extensive necrosis |
Incidence, etiology, and predisposing genetic factors
Bronchial Carcinoids
Bronchial carcinoids account for approximately 2% of all primary lung tumors and roughly 25% of all well differentiated NETs with an incidence rate ranging from 0.2 to 2 per 100,000 per year. Over the past 30 years, the age-adjusted incidence rate of bronchial carcinoids has increased significantly, by approximately 6% per year. This trend may represent the improvement in classification of these tumors, as well as the increase in use of imaging techniques that are able to detect asymptomatic disease. In the United States, there are nearly 6000 new cases per year.
Most carcinoids of the lung are TC, with only 10% to 30% of bronchial carcinoids classified as AC. Patients diagnosed with TC are approximately 10 years younger than those with AC, which occur in the sixth decade of life. In children and adolescents, bronchial carcinoids are the most common primary lung neoplasm. These malignancies are found more commonly in women as compared with men and whites as compared with blacks.
Currently, there are no clearly defined risk factors for bronchial carcinoids. Based on case series, patients with AC are more likely to be smokers as compared with patients with TC. No other carcinogen or environmental exposure is implicated in the development of these malignancies.
Most bronchial carcinoids are sporadic tumors, although rare familial cases have been reported. In less than 5% to 10% of cases, bronchial carcinoids are associated with the autosomal dominant syndrome of multiple endocrine neoplasia type 1 (MEN-1), a rare disorder characterized by a predisposition to neoplasms of the pituitary, parathyroid glands, and pancreas. Furthermore, some sporadic bronchial carcinoid tumors demonstrate inactivation of the MEN-1 gene located on chromosome 11q13. If MEN-1 syndrome is suspected based on family history, an ionized calcium level, intact parathyroid hormone, and prolactin should be sent and the patient should be referred to a geneticist for MEN-1 mutation analysis.
Thymic Carcinoids
Thymic carcinoids are the least common NETs, accounting for approximately 2% to 5% of all thymic tumors and 0.4% of all carcinoid tumors with an incidence rate of approximately 0.2 per 1,000,000 population per year. The largest reported series of thymic carcinoid tumors included 205 patients identified from databases of the International Thymic Malignancy Interest Group and the European Society of Thoracic Surgeons. In this series, the median age was 54 years, the male to female ratio was 3:1, and the majority of patients presented with advanced disease. Tumors were classified as TC, AC, or large cell/small cell neuroendocrine carcinomas in 28%, 40%, and 28% of cases, respectively.
MEN-1 syndrome is associated with up to 25% of thymic carcinoid cases, and approximately 8% of patients with MEN-1 syndrome develop thymic NETs. Because these patients tend to present with advanced disease at diagnosis, prophylactic thymectomy with complete resection of all the anterior mediastinal tissue should be considered, although the benefit of this procedure is controversial. Some experts advocate that males over the age of 25 with MEN-1 syndrome should undergo annual screening to evaluate for thymic NETs; however, there is no consensus on this issue, because there is no study that demonstrates a survival benefit with early diagnosis.
Pathology
Bronchial Carcinoids
The histologic appearance of TC and AC is similar with a uniform population of tumor cells arranged in organoid nests with a moderate amount of cytoplasm with an eosinophilic hue. The finely granular nuclear chromatin frequently has a salt-and-pepper appearance. Necrosis and mitotic activity are used to distinguish TC from AC, as per the WHO criteria detailed herein ( Fig. 1 ). Immunohistochemistry stains for chromogranin A, synaptophysin, and CD56 help to confirm neuroendocrine differentiation. Additional immunohistochemistry stains, including CDX-2, Islet1, TTF-1, or specific hormones and biogenic amines, are useful to separate pulmonary NETs from lung metastasis of well differentiated NETs of other organs.
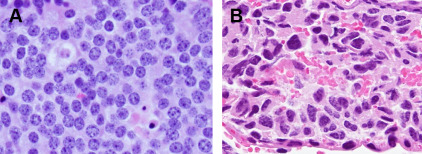
Tumorlets are separated from carcinoid tumors by size, yet the morphology of cells is identical. Nodular neuroendocrine proliferations 0.5 cm or larger are called carcinoid tumors, whereas those measuring less than 0.5 cm are termed tumorlets. Tumorlets are incidental findings in approximately 25% of excised carcinoid tumors and are thought to have no clinical significance, although they can be seen in interstitial or airway inflammatory and fibrosing conditions. Typically no mitoses, no necrosis, and low Ki-67 labeling index are found. A very rare condition called diffuse idiopathic pulmonary neuroendocrine cell hyperplasia is regarded as a preinvasive condition for pulmonary carcinoids. Patients with diffuse idiopathic pulmonary neuroendocrine cell hyperplasia have widespread neuroendocrine cell hyperplasia and tumorlets in their airways, can develop multiple carcinoid tumors, and demonstrate unique findings on computed tomography (CT), with multiple nodules, ground glass attenuation, bronchiectasis, and air trapping owing to small airway obstruction.
Recently, gene copy analysis, genome/exome, and transcriptome sequencing of bronchial carcinoids has been performed. The molecular alterations of bronchial carcinoids are distinct from high-grade NETs in that they have a lower rate of mutations (approximately 0.4 mutations per megabase), lack TP53 and RB1 loss, and contain frequent mutations in chromatin remodeling genes, such as MEN1 , PSIP1 , and ARID1A .
Thymic Carcinoids
For thymic carcinoid tumors, the criteria used to separate TC from AC are the same as those applied to bronchial NETs. These tumors are included in the thymic carcinoma group according to the WHO classification of tumors. Among reported cases of well-differentiated thymic NET, the majority are AC. To make the diagnosis of primary thymic NET, metastasis from another site should be excluded.
The challenge of small biopsies
Although carcinoid tumors can be diagnosed by small biopsies or cytology, it is difficult to separate TC from AC. This distinction usually requires a surgical biopsy or resection specimen. AC may be suspected if mitosis or necrosis is present. In small crushed specimens where mitotic figures are difficult to demonstrate, it may be helpful to use Ki-67 staining because most TC show less than 5% staining, AC are usually 10% to 30%, and most large cell neuroendocrine lung carcinoma or small cell lung cancer (SCLC) usually have a very high proliferation index of 80% to 100%.
Symptoms, approach to making the diagnosis, and staging
Symptoms
Bronchial carcinoids
Two-thirds of bronchial carcinoids develop in the major bronchi. As a result, the most common presenting symptoms include obstructive pneumonia, pleuritic pain, atelectasis, dyspnea, cough, and hemoptysis. Up to 30% of patients with pulmonary carcinoid tumors are asymptomatic at presentation. Carcinoid syndrome (facial flushing, diarrhea, wheezing) is found in 2% to 5% of bronchial carcinoids, most often when liver metastases are present. The syndrome is rare in well-differentiated pulmonary NETs, especially in localized disease, because foregut carcinoids lack aromatic amino acid decarboxylase and cannot make serotonin and its metabolites. Rarely, paraneoplastic syndromes are associated with pulmonary carcinoids ( Table 2 ).
| Syndrome | Symptoms | Biochemical Assessment |
|---|---|---|
| Flushing, diarrhea, and bronchoconstriction; long-term sequelae of cardiac valvular fibrosis (both right and left), fibrosis in the retroperitoneum, and venous telangiectasias | 24-h urine 5-HIAA |
| Central weight gain, striae, hypertension, hyperglycemia, depression, hirsutism |
|
| Swelling and enlargement of the hands, feet, nose, lips, and ears, acrochordon, carpel tunnel syndrome | GH; GHRH; and IGF-1 |
| Anorexia, nausea, muscle aches, myoclonus, ataxia, tremors, altered mental status, seizures, cerebral edema | Urine osmolality, urine sodium, serum sodium |
Thymic carcinoids
Thymic tumors generally occur in the anterior mediastinal compartment, often infiltrating adjacent structures, with approximately 50% of patients presenting with locally invasive disease or mediastinal lymph node metastasis. Symptoms range according to disease extent, from cough, dyspnea, and chest pain to SVC syndrome and hoarseness, secondary to invasion of the recurrent laryngeal nerve. Almost 50% of tumors are complicated by a paraneoplastic endocrinopathy, most commonly Cushing’s syndrome (see Table 2 ).
Diagnosis
Biopsy
Biopsy is necessary for tissue confirmation of carcinoids. Central, bronchial tumors can easily be sampled by bronchoscopy, preferably with flexible bronchoscopy, whereas peripheral lesions can be biopsied by core needle; however, a definitive diagnosis may be difficult to ascertain in small cytology samples, as discussed. For thymic carcinoids, lymphoma or mediastinal germ cell tumor should be ruled out because these diseases are treated medically rather than surgically. Primary resection can be considered for a small, encapsulated thymic mass, along with the thymus, to establish the diagnosis and as definitive treatment. A larger thymic mass with indistinct margins should ideally be biopsied before resection to establish a diagnosis and to determine treatment recommendations. CT-guided core needle biopsy often is performed for the evaluation of thymic masses, although endoscopic or transbronchial ultrasound-guided fine needle aspiration biopsy or mediastinoscopy are helpful to assess the mediastinum.
Imaging
A CT scan of the chest is the recommended imaging for both bronchial and thymic carcinoids, which, in particular, may additionally necessitate mediastinal multiphase CT. Liver and bone metastases are the most common sites of spread. As such, a multiphase CT or MRI with dynamic acquisition and diffuse weighted sequencing of the liver should be used. MRI also can be used to detect and characterize bone metastasis, especially the spine.
Owing to the overexpression of somatostatin receptors, immunoscintigraphy by somatostatin analogs such as octreotide has been approved since 1994 for imaging of patients with NETs. In one series, the sensitivity and specificity were 90% and 83%, respectively. Most octreotide scans are performed with single photon emission CT imaging that enables 3-dimensional scanning or single photon emission CT–CT, which facilitates the display of cross-sectional imaging. However, over the last 15 years, the quality of CT and MRI imaging has improved significantly. In a review that compared modern octreotide scans with CT or MRI scans, multiphase contrast-enhanced CT or MRI scans detected more pathologic lesions than did single photon emission CT–octreotide scanning, and octreotide scans failed to identify any soft tissue lesions or primary tumors that were not seen on CT or MRI scans. These data suggest that routine octreotide scans should not be added to CT scans regularly for surveillance after resection. Furthermore, the need for an octreotide scan preoperatively in patients with resectable disease is debatable, because extrathoracic metastatic disease is rare and cross-sectional imaging with CT scan may be better for detecting such lesions. Baseline octreotide scans are useful in patients with carcinoid syndrome or metastatic disease to determine somatostatin receptor status and the therapeutic utility of somatostatin analogs.
Fluorine 18-fluorodeoxyglucose (FDG) PET scanning may be less accurate because these indolent tumors generally have a low standard uptake value, although some authors still advocate for its potential use. Recent series have shown that FDG-PET is useful for the assessment of intermediate- and high-grade NETs and may have prognostic value. In the preoperative setting, the need for a PET scan is debatable as demonstrated by the findings of a recent, large retrospective study where the sensitivity of 18F-FDG PET/CT for mediastinal lymph node metastasis was considerably poorer for patients with bronchial carcinoid than that for non-SCLC (NSCLC; 33% vs 84% for NSCLC), but the specificity was comparable (94% vs 89% for NSCLC), suggesting that lymph node metastases accurately cannot be ruled out with a negative PET/CT. As such, in the preoperative setting, if treatment decisions are based on N2 status, mediastinal staging is required with either endobronchial ultrasonography or mediastinoscopy.
Echocardiogram should be performed at diagnosis and during follow-up for patients with carcinoid syndrome to evaluate for the presence of carcinoid heart disease, which is characterized by plaquelike deposits of fibrous tissue in the right and left valves and endocardium and occurs in more than 50% of patients with carcinoid syndrome.
Biochemical assessment
Increased chromogranin A levels have been measured in serum or plasma, although they have been found to be lower in pulmonary carcinoids compared with gastroenteropancreatic NETs. In the setting of advanced or metastatic disease, chromogranin A levels can be useful to follow disease activity. Although rare in bronchial and thymic carcinoids, if carcinoid syndrome is suspected, a 24-hour urinary excretion of 5-hydroxyindoleacetic acid should be measured (see Table 2 ). Serotonin levels should not be followed because there are several assays with variable sensitivity and specificity, and levels can be affected by release of platelet serotonin and tryptophan and serotonin-rich foods. Patients with sporadic, non–MEN-1–associated thymic carcinoids should undergo evaluation for Cushing’s syndrome, because its incidence is high in this population. Acromegaly and hyponatremia secondary to syndrome of inappropriate antidiuretic hormone are rare and usually are seen only in sporadic cases (see Table 2 ).
Staging and Prognosis
Bronchial carcinoids
Bronchial carcinoid tumors are staged according to the TNM classification used for NSCLC in the seventh edition of the American Joint Committee on Cancer staging system. The International Association for the Study of Lung Cancer proposed and approved this staging in 2009 when it was determined that the TNM staging system was helpful in predicting prognosis for bronchial carcinoids. Applying this staging system to cases in the National Cancer Institute Surveillance, Epidemiology, and End Results registry and cases submitted to the International Association for the Study of Lung Cancer database, it was determined that 5-year overall survival (OS) for patients with stage I, II, III, and IV disease were 93%, 74% to 85%, 67% to 75%, and 57%, respectively.
Survival is significantly better for TC than for AC. The 5- and 10-year survival rates have been reported as 87% to 100% and 82% to 87%, respectively, for TC, and 30% to 90% and 35% to 56%, respectively, for AC. Other predictors of survival include tumor size, nodal involvement, higher mitotic rates (ie, atypical subtype), and age older than 60. Importantly, patients with multiple nodules have a very favorable prognosis, likely because these individuals tend to have the underlying preinvasive lesion, diffuse idiopathic pulmonary neuroendocrine cell hyperplasia, and multiple synchronous primaries rather than intrapulmonary metastasis.
Thymic carcinoids
A formal staging classification has not been developed for thymic carcinoids and therefore Masaoka-Koga system, Masaoka system, or the terms “local,” “locally advanced,” and “metastatic” are used commonly ( Table 3 ). Thymic carcinoids are felt to have a more aggressive behavior, recur locally, and metastasize more frequently, as compared with other NETs arising elsewhere. In the largest series of thymic carcinoids to date, median survival was 13.5 years for Masaoka stages I and II, 7.3 years for stage II, 3.8 years for stage IVa, and 4.2 years for stage IVb. Masaoka stage and R0 resection was a significant prognostic factor for survival, although histologic subtype did not show any effect on survival. Another study did show that histologic subtype effects survival. In a group of 50 patients with long-term clinical follow-up, disease-free survival correlated with tumor grade; low-grade tumors had 5- and 10-year disease-free survivals of 50% and 9%, intermediate-grade tumors had a disease-free survival of 20% at 5 years, and none were disease free at 10 years. None of the patients with high-grade tumors were disease free at 5 years.
| Stage | Description |
|---|---|
| I | Macroscopically encapsulated with no microscopically detectable capsular invasion |
| II | Macroscopic invasion of mediastinal fatty tissue or mediastinal pleura or microscopic invasion into the capsule |
| III | Macroscopic invasion into surrounding structures (pericardium, great vessels, lung) |
| IVa | Pleural or pericardial dissemination |
| IVb | Lymphogenous or hematogenous metastasis |
Treatment of bronchial carcinoids
Stages I, II, and III
Surgery
Surgery is the primary treatment modality and the best curative option for patients with bronchial carcinoids. Because carcinoids often present centrally, pneumonectomy or bilobectomy are used frequently. However, lung parenchymal-sparing surgery such as bronchial sleeve, or wedge resection is favored in view of the low recurrence potential, especially for TC. Because carcinoids generally do not spread submucosally, a surgical margin as close as 5 mm is considerate adequate. If positive margins are reported and the patient can tolerate a repeat surgery, repeat resection with wider margins can be considered. For patients with favorable prognostic features, such as typical histology and absence of lymph node involvement, a more limited resection has been proposed. Patients with AC should be resected using the same principles guiding surgery for NSCLC.
Because mediastinal lymph node metastases occur in up to 20% of cases of TC and 30% to 70% of cases of AC, complete mediastinal lymph node dissection is advocated, with surgical resection of nodal metastases when feasible. Multiple series have found decreased incidence of local recurrence and improved survival when complete mediastinal lymph node dissection is performed.
Adjuvant therapy
Currently, there is no consensus on adjuvant therapy in bronchial carcinoids after resection; trials in the adjuvant setting are lacking. For this reason, it is important to establish appropriate management and treatment plans within a multidisciplinary tumor board. In general, adjuvant chemotherapy after surgical resection for patients with TC with or without regional lymph node metastases (stages I, II, and III) is not recommended because the risk of recurrence has been shown to be low. In a series of 291 resected TC, only 3% of patients developed recurrence. Similarly, after surgical resection, patients with stage I AC are followed expectantly. However, because systemic recurrence occurs more frequently in patients with AC with N1 or N2 involvement (stages II and III disease), adjuvant chemotherapy has been advocated by some based on retrospective literature. Nonetheless, the optimal adjuvant regimen in this setting currently is unknown. Owing to the similarities with SCLC, etoposide and cisplatin or carboplatin generally are used. Although the benefit is uncertain, multiple small retrospective reviews demonstrate that AC patients with N1 or N2 involvement who receive adjuvant platinum-based therapies still experience distal recurrences and die of disease within 10 years of treatment. Prospective clinical trials in this setting are clearly needed to determine the best regimen for adjuvant treatment to reduce recurrence rates and improve OS.
Radiation therapy
The use of radiation therapy for carcinoid tumors is most similar to its pattern of use in NSCLC, although there is a lack of data and uncertainty of benefit. For tumors that are resectable, adjuvant radiation therapy may be offered in situations of residual disease (R1 resection) and mediastinal lymphadenopathy (N2 disease). The use of adjuvant radiation therapy for nodal disease is probably of greater utility in the more aggressive AC. However, carcinoid tumors generally are thought to be less responsive to radiation therapy than SCLC.
Locally Advanced Unresectable Disease
Definitive radiation therapy can provide effective treatment for a locally unresectable primary tumor. The addition of chemotherapy, such as a platinum-based regimen used in SCLC, is used occasionally, although response rates seem lower; whether this approach improves results as compared with radiation therapy alone remains uncertain. Data to guide optimal treatment are limited secondary to the rarity of these tumors and lack of prospective clinical trials.
Surveillance of Resected Bronchial Carcinoids
Local and distant disease recurrence can occur years after surgical resection and, for this reason, long-term follow-up of patients with bronchial carcinoids is warranted. The optimal surveillance strategy is not defined currently. The National Comprehensive Cancer Network guidelines recommend reevaluation every 3 to 12 months after resection and then every 6 to 12 months for up to 10 years. However, this may not be necessary in patients with node-negative TC, because recurrence is rare in this population. Somatostatin receptor scintigraphy is not recommended routinely during surveillance after curative resection unless metastatic disease is suspected. Biochemical evaluations with chromogranin A and 5-hydroxyindoleacetic acid may also be helpful to monitor disease activity in the setting of metastatic disease, but do not need to be monitored postoperatively.
Stage IV Disease
Overview
Because carcinoid tumors can be relatively indolent and there are no curative therapeutic options in the metastatic setting, quality of life is a critical consideration in deciding on a treatment plan. In asymptomatic patients with TC and AC and a low tumor burden, a watch and wait policy with regular follow-up and imaging every 3 to 6 months can be pursued. Key factors in deciding when to initiate treatment include how quickly the patient progresses off treatment, the burden of disease, and the presence of hormone-related symptoms. In patients who have clinically significant tumor burden and/or carcinoid syndrome, somatostatin analogs as first-line treatment should be considered, in the setting of a positive octreotide scan. Chemotherapy or targeted therapy, such as everolimus, should be considered for those patients with more rapidly progressing tumors or those who have progressed on less toxic treatments. Additional prospective, randomized studies are needed for both traditional cytotoxic and molecularly targeted agents because treatment principles generally are extrapolated from the experience with the more common gastrointestinal carcinoids. Palliative local therapy such as local resection, radiation therapy, radioablation, or cryoablation can be used for symptomatic lesions.
Somatostatin analogs
Retrospective reviews including small numbers of patients with pulmonary carcinoids have reported on the use of somatostatin analogs with improvement in carcinoid syndrome symptoms, as well as prolonged disease control and survival, with very few individuals achieving a tumor response. These results are similar to the experience with octreotide in gastrointestinal carcinoids. In the Placebo Controlled, Double Blind, Prospective, Randomized Study on the Effect of Octreotide LAR in the Control of Tumor Growth in Patients with Metastatic Neuroendocrine Midgut Tumors (PROMID) study, long-acting release (LAR) octreotide acetate significantly prolonged time to tumor progression compared with placebo in patients with newly diagnosed functionally active or inactive well-differentiated midgut NETs (14.3 vs 6 months; hazard ratio [HR], 0.34; 95% CI, 0.20–0.59; P = .000072) without an improvement in OS. Additionally, in the phase III, randomized, placebo-controlled Controlled Study of Lanreotide Antiproliferative Response In NeuroEndocrine Tumors (CLARINET) study, lanreotide autogel significantly prolonged time to tumor progression compared with placebo in 204 patients with nonfunctional enteropancreatic NETs (median not reached versus a median of 18 months; HR, 0.47; 95% CI, 0.30–0.73; P <.001). Neither of these studies have shown a benefit in OS, likely owing to the high rate of somatostatin analog use in each of the placebo arms after progression.
Although neither of the prospective studies included foregut NETs, somatostatin analogs should be considered as first-line systemic treatment for patients with advanced bronchial carcinoids of low proliferative index, who have positive octreotide scans, owing to their excellent safety profile. Caution should be used in patients with lesions that have a high mitotic index, a high tumor burden, or rapidly progressing disease; and first imaging should be performed relatively soon after administration of the somatostatin analog to monitor response to therapy.
Peptide receptor radionuclide therapy
In patients with advanced disease that is resistant to octreotide, peptide receptor radionuclide therapy with radiolabeled somatostatin analogs is being evaluated in several centers. Indium-111, yttrium-90, and lutetium-177 are linked to somatostatin analogs and then internalized by tumor cells. [90Y-DOTA]-DPhe1-Tyr3-octreotide (yttrium-90 DOTATOC) and lutetium-177 DOTATOC show particular promise in selected patients. An early phase II study of yttrium-90 DOTATOC found the response rate to be as high as 29% in 7 bronchial carcinoids. In a large retrospective study of 1109 metastatic NETs, which included 84 bronchial carcinoids, a 29% partial response and median survival of 40 months from diagnosis were demonstrated. Grade 3 to 4 adverse events were reported in 13% of patients, including mainly renal or hematologic toxicities. Although these are promising results, peptide receptor radionuclide therapy remains experimental, at least in the United States, and can be only be administered in the setting of a clinical trial.
Cytotoxic therapy
Data regarding the efficacy of chemotherapy specifically in bronchial carcinoid (as opposed to gastrointestinal carcinoids) are lacking, because this tumor type has not been studied independent of other NETs and has been omitted occasionally from such trials. Furthermore, many of the older studies have used outdated classification systems for carcinoids and different criteria for response. Various chemotherapeutic agents have been used, including doxorubicin, 5-fluorouracil, dacarbazine, cisplatin, carboplatin, etoposide, streptozocin, and interferon-alpha. Newer agents are being studied actively in neuroendocrine carcinoma. Results from larger retrospective and prospective studies are summarized in Table 4 .
Related posts:
 Neuroendocrine Tumors—Current and Future Clinical Advances
Neuroendocrine Tumors—Current and Future Clinical Advances
 Surgical Treatment of Small Bowel Neuroendocrine Tumors
Role of Somatostatin Analogues in the Treatment of Neuroendocrine Tumors
Surgical Treatment of Small Bowel Neuroendocrine Tumors
Role of Somatostatin Analogues in the Treatment of Neuroendocrine Tumors
 Neuroendocrine Tumors—Current and Future Clinical Advances
Systemic Therapies for Advanced Pancreatic Neuroendocrine Tumors
Neuroendocrine Tumors—Current and Future Clinical Advances
Systemic Therapies for Advanced Pancreatic Neuroendocrine Tumors
 Surgical Management of Pancreatic Neuroendocrine Tumors
Surgical Management of Pancreatic Neuroendocrine Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


