Fig. 16.1
Proportion of major bone sarcoma groups by age (SEER, 2000–2011)
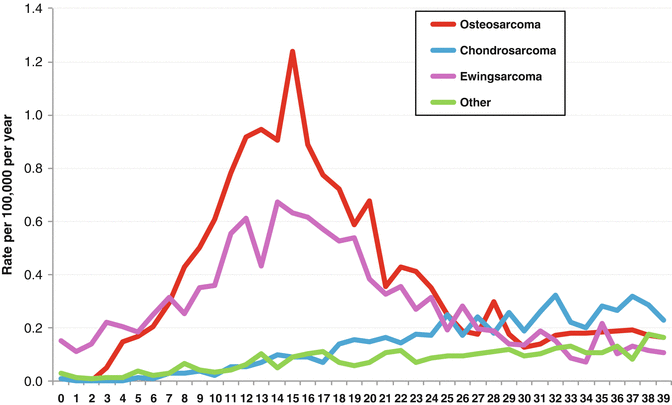
Fig. 16.2
Bone incidence by type and age from SEER (2000–2011)
The clinical significance of bone sarcomas in adolescents and young adult populations lies in four major aspects. First, the treatment of bone sarcomas is complex, multidisciplinary and intensive. While osteosarcoma and Ewing sarcoma are treated with surgery and chemotherapy, chondrosarcomas are generally regarded as refractory to chemotherapy and radiotherapy and are treated by surgery. Second, bone sarcomas contribute a disproportionate burden of morbidity and lethality attributable to cancers occurring in this group. Third, particularly for Ewing sarcoma, bone sarcomas are treated at both paediatric and adult institutions, and age-dependent differences in overall survival have been demonstrated. Finally, the morbidity and excess mortality associated with survivors of bone cancer in the AYA group are poorly understood, but likely highly significant contributors to the community burden of these diseases.
16.2 Osteosarcoma
16.2.1 Epidemiology and Aetiology
Osteosarcoma arises as a consequence of the malignant transformation of cells of the osteoblast lineage. The age-adjusted incidence is 0.33/100,000 population, and the peak incidence is in adolescence and young adulthood, although there is a second peak in old age. Males outnumber females in the AYA range by 1.4:1. The first peak coincides with the onset of peak bone growth and its aftermath, while the second peak occurs in association with environmental exposures like radiation or in association with increased states of bone turnover, like Paget’s disease of the bone. These features suggest that osteoblast proliferation is a factor in the development of osteosarcoma. Other benign predisposing lesions associated with osteosarcoma include fibrous dysplasia, bone infarction, chronic osteomyelitis, giant cell tumour of the bone and osteogenesis imperfecta [10]. The most common sites of disease are the long bones of the lower limb (68 %), the upper limb (10 %) and the skull (9 %; Fig. 16.3).
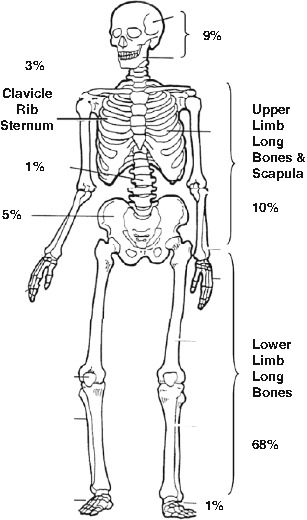
Fig. 16.3
Skeletal location of osteosarcoma primary tumours
There are both environmental and genetic risk factors for the development of osteosarcoma. The strongest known environmental risk factor is prior exposure to radiation, accounting for less than 5 % of all cases, but particularly in older patients [4]. The risk of radiation-induced osteosarcoma is increased in the context of heritable syndromes associated with osteosarcoma. The best known of these is the retinoblastoma syndrome, in which the risk of osteosarcoma is greatly increased by exposure to ionising radiation [22]. Other heritable syndromes associated with osteosarcomas include the Li-Fraumeni syndrome, associated with germline mutations in TP53; and the helicase syndromes, which are associated with developmental features (Bloom syndrome, associated with mutations in BLM; Rothmund-Thomson syndrome, associated with mutations in RECQL4; and Werner syndrome, associated with mutations in WRN) [34]. Importantly, the likelihood of genetic factors contributing to cancer risk is greater in younger patients and may have clinical implications for choice of therapy. Taking a good family history is therefore important in the assessment of AYA with any sarcoma.
16.2.2 Pathology and Biology
There are several well-described histologic varieties of osteosarcoma, but they are usefully divided into high-grade and low-grade tumours. The most common subtype is high-grade central osteosarcoma, which arises typically as a large, metaphyseal lesion of the long bones with intramedullary extension. Histologically, central high-grade osteosarcoma comprises malignant osteoblasts associated with neoplastic osteoid (disorganised in architecture). Further subcategorisation recognises osteoblastic, fibroblastic, chondroblastic and giant cell-rich variants, without clear clinical associations. High-grade central osteosarcoma is characterised by complex, unstable and aneuploid genotypes. Telangiectatic osteosarcoma is a high-grade tumour characterised by large, vascular and hemorrhagic spaces, typically arising within the metaphyses of the long bones. They account for less than 4 % of all osteosarcomas. Small cell osteosarcoma account for less than 2 % of all osteosarcomas and are characterised by small cells with lacelike malignant osteoid production. The main differential diagnosis for small cell osteosarcomas is Ewing sarcoma. Periosteal and high-grade surface osteosarcomas are tumours arising from the bone surface and comprise less than 5 % of all osteosarcoma. All of these high-grade lesions are typically treated with chemotherapy as well as surgery. Distinction between periosteal osteosarcoma, usefully regarded as of intermediate grade and with a lower metastatic potential, and high-grade surface osteosarcoma which behaves akin to high-grade central osteosarcoma is sometimes challenging. The value of chemotherapy for periosteal osteosarcoma is doubtful [15].
Low-grade central and parosteal types account for 1–2 % of all osteosarcoma and connote a good prognosis. They arise either as central or surface lesions and are characterised by relatively well-differentiated cells within osteoid that resembles fibrous dysplasia. Genetically, they differ from the high-grade bone tumours in being cytogenetically stable and relatively diploid and are typically associated with high-level amplification of MDM2 and CDK4 (2013b). The outcomes for this group of osteosarcomas are excellent, with 5-year survival rates in excess of 90 %. They are typically treated with surgery alone, unless there is evidence of de-differentiation.
16.2.3 Diagnosis and Staging
Clinically, osteosarcoma typically presents as unremitting pain that is worse at night and swelling and deformity. In some cases, patients present with pathologic fracture. The initial diagnosis is often made on plain radiology, which typically shows anatomic distortion and bony sclerosis or lucency, with or without evidence of fracture. As a general rule, if a diagnosis of osteosarcoma is suspected on plain radiology, the patient should be referred to an experienced orthopaedic oncologist working in a multidisciplinary team. This is an important point for adolescent and young adults, who—with in contrast with younger children—may be diagnosed outside specialised sarcoma centres. Histologic confirmation of the diagnosis is made either on open or core biopsy. It is important that the biopsy is performed with the awareness of the possible diagnosis, since the biopsy site and track can influence subsequent surgical planning.
Staging should involve comprehensive CT and MR imaging of the entire affected bone, since osteosarcoma is not infrequently associated with skip metastases. These are metastatic deposits that are located within the bone in which the osteosarcoma arises, but are not contiguous with the primary site. They connote an adverse outcome. In addition, a CT scan of the chest is also indicated, as the chest is the most common site of metastatic deposits. Most major referral centres may also perform a PET scan or technetium scintigraphy to identify occult bony metastases, since the second most common site of metastatic disease is to other bony sites. The outcomes for osteosarcoma that is metastatic at diagnosis are very poor.
16.2.4 Treatment
The introduction of adjuvant chemotherapy more than tripled survival for non-metastatic osteosarcoma [18]. Surgery remains critical to cure, and radiotherapy is infrequently used for local therapy, either alone or as an adjuvant. For non-metastatic, high-grade osteosarcoma, treatment most commonly involves preoperative chemotherapy with a backbone that involves doxorubicin, cisplatin and methotrexate (MAP). There has been debate as to the value of methotrexate, but it is included in most current regimens in young patients [6]. Regimens including this triad yield 5-year survival rates in excess of 70 % for patients with resectable disease. Recent clinical trials have tested the role of adjuvant mifamurtide (muramyl tripeptide), postsurgical response-adapted therapy in poor responders to neoadjuvant (MAP) and adjuvant interferon in good responders. Over 660 patients with localised osteosarcoma were treated preoperatively with MAP and then randomly assigned to receive ifosfamide and/or mifamurtide. The addition of mifamurtide without ifosfamide significantly improved 6-year OS from 70 to 78 % (P = 0.03) [27]. Unexpectedly, this effect was not observed in the arm containing ifosfamide, perhaps due to an unexplained drug interaction. Mifamurtide is currently only approved for use in the EU and other countries including Mexico, Libya, Ukraine, Venezuela, Israel and South Korea but not in North America.
After metastasis at diagnosis, the most powerful known prognostic factor for overall survival in patients with osteosarcoma is response to neoadjuvant chemotherapy. The 5-year overall survival for good responders is over 80 % compared to 52 % for those with a poor response [6]. There appears to be a minor effect of age of onset on outcomes following treatment of resectable osteosarcoma. This is perhaps best illustrated in a study which pooled data on 4,838 patients from several study groups in which adolescents and adults appeared to have slightly worse outcomes than for children (hazard ratio 1.14, P = 0.03; [6]. Interestingly, most of the difference appears linked to gender, with males having worse responses than for females (HR 0.85, P = 0.003). A smaller analysis of COG trials reported a much stronger effect of age and weaker influence of gender but did not analyse gender, age and toxicity as in the Collins study [20]. It is perhaps worth noting that these are ‘clinical trial’ populations and in an unselected population registry-based studies the influence of increasing age is clearly apparent [8, 36]. The EURAMOS-1 trial, conducted as a transatlantic collaboration between the Northern American and European co-operative trial groups, randomised patients following neoadjuvant MAP and surgery to either a good response arm or a poor response arm. The good response arm demonstrated more than 90 % necrosis of tumour cells in the postoperative specimen, where the poor response arm demonstrated less than 90 % necrosis. In the EURAMOS-1 trial, good responders were randomised to receive a year of adjuvant pegylated interferon-alpha or to continue with MAP. Recently published results suggest no benefit for the interferon arm [2]. Those in the poor response arm were randomised to receive a regimen containing ifosfamide and etoposide or further MAP. The initial results have been reported in abstract form to show no benefit and increased toxicity for the arm containing ifosfamide and etoposide [25]. This unexpected and important result suggests that, although response to preoperative chemotherapy is a powerful prognostic indicator, adaptation of postoperative chemotherapy as tested in EURAMOS-1 does not alter outcomes.
Outcomes for metastatic osteosarcoma at diagnosis, or following early relapse, remain poor (5-year overall survival for those with metastases at diagnosis 26 % compared to 63 % for those without metastases, P < 0.001; [6]). However, it is important to note that oligometastatic disease to the lungs that occurs more than 12 months following definitive therapy can be managed using metastasectomy with curative intent [13, 26].
16.2.5 Late Effects
An institutional cohort of 883 evaluable patients with non-metastatic osteosarcoma, with a median age at diagnosis of 15 years, was followed for a median of over 10 years, with 310 deaths [24]. The major causes of death were recurrent osteosarcoma (22 % of deaths from recurrent osteosarcoma occurred after 10 years of follow-up), second malignant neoplasms (5 %) and cardiac impairment (2 %). Overall, 36 survivors (6 %) developed a second neoplasm, after a median interval of 8 years. For a median of 9.5 years from diagnosis, fertility was impaired in 47/54 evaluated male patients (31/44 post-pubertal patients azoospermic) and was probably increased by cumulative ifosfamide doses. It is likely that these patients were selected on the basis of clinical infertility and do not represent the rates amongst all male survivors. Amongst female survivors, 6/207 (2.8 %) experienced permanent amenorrhoea, most of whom were over 35 years of age at diagnosis. Since this survivorship cohort had a median age in their twenties, long-term late effects will undoubtedly continue to accumulate, probably varying on the basis of treatment regimen and patient factors including genetic predisposition to cancer. The consequences of surgery are likely significant, and rehabilitation programmes are an important part of management of this disease.
16.3 Ewing Sarcoma
16.3.1 Epidemiology and Aetiology
Ewing sarcoma is a small round cell sarcoma of uncertain histogenesis, but probably arising in a mesenchymal stem cell [35]. Ewing sarcoma (ES) typically arises in the bone, but may arise within any tissue. Most ES are characterised by recurrent translocations involving EWSR1 and a number of partner genes. The age-adjusted incidence of Ewing sarcoma is 0.2/100,000 of the population, and 80 % of cases arise under 20 years of age [37]. Ewing sarcoma does occur in individuals over 30 years of age, but this is rare. There is a slight male-female preponderance (1.4:1). There are no known environmental associations. Although neither familial clustering nor a genetic basis for ES has been identified, EW is more common in Caucasian than African Americans [40], and atypical inheritance patterns have been observed that depart from Mendelian models [31].
ES is a cancer in which there is a strong age-dependent effect on survival [14, 21]. Children have a markedly better survival from ES than adolescents and young adults. This effect is at least partly independent of patterns of care, since on the same randomised trial, the event-free survival for those over 17 years of age was 44 % compared to 70 % for those under 10 years of age (relative risk 2.5, P = 0.001) [14]. Gender may also play a role in determining outcomes for older patients, with males having worse survival than females [21].
16.3.2 Pathology and Biology
ES typically occurs in the diaphyseal regions of the long bones, pelvis or ribs, but may occur at almost any anatomical site. The site of disease is important prognostically, since outcomes are worse for axial compared to appendicular tumours [14]. Most cases comprise homogenous, small, round cells with scant cytoplasm and usually little matrix (2013b). They are typically CD99 (MIC2) positive immunohistochemically, although this marker is neither always present nor specific for ES. The diagnosis is typically made on detection by fluorescence in situ hybridisation or PCR of rearrangements involving EWSR1 that result in fusion genes. The most common partner is the transcription factor FLI1 (85 %), but Ets family members including ERG, ETV1, FEV and others have been described. ES bears a low burden of mutations, including in the canonical cancer genes, and evidence is gathering that the fusion gene may function epigenetically [36]. A recent finding of interest has been the exquisite sensitivity of ES cell lines to poly ADP-ribose phosphate inhibitors [12], although the basis for this is not understood (Fig. 16.4).
 Fertility Preservation in the Pediatric Setting
Fertility Preservation in the Pediatric Setting
 Economic Evaluation in Adolescent and Young Adult Cancer: Methodological Considerations and the State of the Science
Economic Evaluation in Adolescent and Young Adult Cancer: Methodological Considerations and the State of the Science
 Colorectal and Anal Tumors
Colorectal and Anal Tumors
 Promoting Health and Care Transitions in the Long-Term AYA Survivor
Promoting Health and Care Transitions in the Long-Term AYA Survivor
 Acute Lymphoblastic Leukemia
Acute Lymphoblastic Leukemia
 Cancer of the Kidney, Bladder, and Prostate
Cancer of the Kidney, Bladder, and Prostate




Related posts:
Fertility Preservation in the Pediatric Setting
Colorectal and Anal Tumors
Promoting Health and Care Transitions in the Long-Term AYA Survivor
Acute Lymphoblastic Leukemia
Cancer of the Kidney, Bladder, and Prostate
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
