Bone Sarcomas
Jean-Yves Blay
Olivia Bally
I. INCIDENCE AND NOSOLOGY
Bone sarcomas account for about 10% of all sarcomas, and, therefore, 0.1% to 0.2% of all malignant tumors in adult patients. They make up 5% to 10% of malignant tumors in children, and 30% to 50% of bone sarcomas arise in children.1,2 The histologic subtypes of bone sarcomas are less heterogenous than those of soft tissue sarcomas, yet these tumors show very heterogenous behavior and require very different treatments.
Osteogenic sarcoma (OS), chondrosarcoma, Ewing sarcoma (ES), and a composite group of rarer histologic subtypes (e.g., undifferentiated pleomorphic sarcoma, leiomyosarcoma) represent the most common histologic subtypes of bone sarcomas. The term osteosarcoma refers to OS of the bone, that is, a malignant tumor of the bone, producing osteoid matrix. OS is the most frequent primary tumor of bone with an incidence of 0.2 to 0.3/100,000/year. The incidence is higher in adolescents and peaks at the age of 15 to 19. OS arise most often from the limbs, specifically in the superior extremity of the tibia and lower extremity of the femur, but can be observed in any bone site, including the toes and head and neck.
Chondrosarcoma is the most common bone sarcoma of adults, with an incidence of close to 0.2/100,000/year. The age at diagnosis is most often between 30 and 60. Chondrosarcomas arise most often from the pelvic or shoulder girdle, and from flat bones, including the skull, but can also arise from any other bone site.
ES is the third most common bone sarcoma, occurring most frequently in children and teenagers with a median age of 15 years at diagnosis, and in about 30% of cases in adults, up to 80 years. Together with primitive neuroectodermal tumors, they constitute the Ewing Family of Tumors (EFT), defined by a specific set of translocations, the most frequent being the t(11;22) chromosome translocation that results in the EWS-FLI-1 gene rearrangement. Members of the EFT should be regarded as the same tumor. ES may arise from any bone, and soft tissues as well, in 30% of the cases, and its treatment in both cases is similar. The pelvic bones, thoracic wall, and vertebrae are frequent primary sites.
A small subgroup of patients present with primary sarcoma of the bone with different histologic subtypes. Most of these were designated as malignant fibrous histiocytosarcoma (MFH) of the bone, but are now categorized more often as undifferentiated pleomorphic sarcoma of the bone or bone leiomyosarcoma. They tend to occur at an adult
age, and often affect the long bones, with a clinical presentation close to that of osteosarcoma of the bones. They are usually treated with osteosarcoma protocols, but have distinct age distributions and clinical behavior.
age, and often affect the long bones, with a clinical presentation close to that of osteosarcoma of the bones. They are usually treated with osteosarcoma protocols, but have distinct age distributions and clinical behavior.
Chordoma is another rare malignant tumor of the bone, arising from remnants of the notochord mostly in the sacrum, vertebrae, and from the base of the skull, with an incidence of 0.1/100,000. The median age at diagnosis is 60, with skull-base presentations generally affecting a younger population, including children.
Giant cell tumors of the bone are most often locally aggressive tumors arising from the long bone of young adults, with frequent local relapses, and occasional distant metastases in 15% to 20% of patients. Giant cell tumors of the bone have an incidence of close to 0.2/100.000/year.
There are several other rarer malignant bone tumors, such as adamantinoma. It is important to remember, however, for differential diagnosis, that the first cause of bone tumors is secondary cancers, which should be investigated thoroughly, particularly in adult patients aged above 50.
II. PREDISPOSING FACTORS FOR BONE SARCOMA
Several risk factors for the development of bone sarcoma have been identified. These include genetic predispositions, predisposing diseases, and previous radiation therapy.2
There are a variety of genetic familial predispositions associated with an increased risk of cancer. Li-Fraumeni syndrome is caused by a mutation of a tumor suppressor gene TP53. Patients with this rare syndrome are more prone to developing a variety of cancers, including bone and soft tissue sarcomas. Hereditary retinoblastoma is caused by a mutation in the RB1 gene. Affected patients are prone to developing bilateral retinoblastoma and have an increased risk of developing bone or soft tissue sarcomas, including osteosarcoma or leiomyosarcoma at adolescence or in early adulthood. Bessel-Hagen disease, also known as hereditary multiple exostosis, is a rare inherited skeletal disorder, caused by mutations of the EXT1 or EXT2 genes, which causes short stature and deformities of the limbs and extremities. Each osteochondroma can potentially develop into a malignant bone sarcoma, most often chondrosarcoma. Ollier disease, or multiple enchondroma, is also a rare disease often caused by mutations of IDH1 and IDH2 genes. OD has a wide variety of clinical presentations. The multiple enchondromas characteristic of this disease have a risk of developing into a bone sarcoma (most often, a chondrosarcoma).
Paget disease of bone is a benign disorder characterized by increased bone resorption accompanied by aberrant new bone formation. Bone sarcomas (mostly osteosarcomas) develop in about 1% of people with Paget disease, usually when many bones are affected. It affects mostly people older than 50. The prognosis of these tumors
is generally poor. The incidence of transformation seems to have decreased since the introduction of bisphosphonates for the treatment of Paget disease.
is generally poor. The incidence of transformation seems to have decreased since the introduction of bisphosphonates for the treatment of Paget disease.
Radiotherapy increases the risk of bone sarcomas. Sarcomas can arise following exposure to radiation given to treat other cancers in the area. The average time between radiation exposure and diagnosis of a bone sarcoma is about 10 years. Radiation exposure is, however, a very rare cause of bone sarcomas.
III. CLINICAL SYMPTOMS AND DIAGNOSIS OF BONE SARCOMAS
Because of their rarity and the complexity of histologic classification as well as of multimodal treatment, all patients with a suspected primary bone sarcoma should be referred to a specialized reference center with expertise in the treatment of primary bone tumors, involving dedicated pathologists, radiologists, orthopedic surgeons, radiation oncologists, medical oncologists, and pediatric oncologists.1,2
Symptoms of bone sarcomas depend on the size and location of the tumor. The most common symptom is bone pain, progressively increasing, at night, together with swelling, and functional impairment. Pathologic fractures can be the first symptom. Nerve or vascular compression is less frequent. Less common also are fever, unexplained weight loss, fatigue or anemia.
After a clinical examination, bone X-rays will generally be proposed: bone destruction, cortical fracture, and abnormal osteogenesis in the soft part are the features of bone tumors (Fig. 18.1). Often, the radiologic presentation enables one to recognize a bone tumor such as OS. Magnetic resonance imaging (MRI) of the whole anatomical compartment with adjacent joints is required for the diagnosis of extremities and pelvic tumors, and enables the clinician to assess the size and spread in the bones or in the surrounding soft tissues. Computerized tomography scan (CT scan) is recommended for the precise assessment of the primary bone tumor of the trunk and head and neck. Thoracic, abdominal, or pelvic CT scan is also an essential part of the workup for staging, enabling the clinician to assess the metastatic spread of the sarcoma in the lungs and beyond. Positron emission tomography (PET scan) with F18 glucose and bone scintigraphy with Tc99 are also frequently used to assess the tumor spread and to analyze bone remodeling and metabolic activity.
Biopsy must be performed by an experienced radiologist or surgeon after multidisciplinary assessment of the clinical and radiologic file, to define the biopsy track (which will be subsequently removed en bloc). Core needle biopsies or incisional/excisional biopsies can be performed, depending on the presentation. The histopathologic assessment of the tumor must be performed by an experienced pathologist.1,2 The determination of the molecular alteration of the tumor, for example, the presence of a fusion transcript EWS-FLI-1, or of the t(11,22) translocation for ES is of growing importance for the management of
bone sarcomas, as it is for soft tissue sarcomas as well. Grading of the sarcoma is also a key parameter in selecting the treatment, and should be determined by an experienced pathologist. Blood samples are part of the workup to evaluate the function of the liver, kidneys, and blood cells. An increase in blood levels of alkaline phosphatase and lactate dehydrogenase is often detected in bone sarcoma, and may reflect the importance of tumor mass, and have prognostic value.
bone sarcomas, as it is for soft tissue sarcomas as well. Grading of the sarcoma is also a key parameter in selecting the treatment, and should be determined by an experienced pathologist. Blood samples are part of the workup to evaluate the function of the liver, kidneys, and blood cells. An increase in blood levels of alkaline phosphatase and lactate dehydrogenase is often detected in bone sarcoma, and may reflect the importance of tumor mass, and have prognostic value.
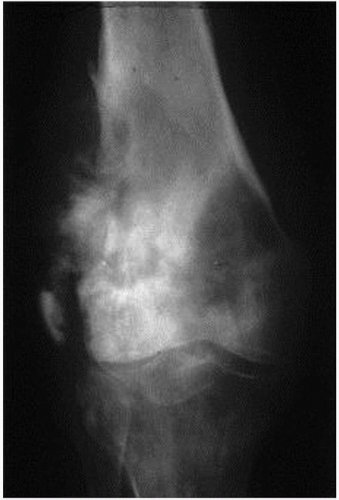 FIGURE 18.1 Osteosarcoma of the lower extremity of the femur. |
After the workup, the histologic subtype of the bone tumors as well as the stage of the tumor will be established, enabling the determination of a therapeutic strategy and refinement of the prognosis.
IV. STAGING
Bone tumors are staged according to the American Joint Committee on Cancer (AJCC) criteria as well as the criteria of the Musculoskeletal Tumor Society.
The criteria in the AJCC staging system are tumor grade, tumor size, and presence and sites of metastases. There are four tumor grades:
Grade 1: Well differentiated—low grade
Grade 2: Moderately differentiated—low grade
Grade 3: Poorly differentiated—high grade
Grade 4: Undifferentiated—high grade
Tumors are divided into size categories of 8 cm or more. Tumor size determines A and B, substages of stages I and II, and stage III.
T1 = ≤8 cm
T2 = >8 cm
T3 = Discontinuous tumors in the primary bone site
Metastatic status is subdivided by presence and location of metastases:
M0 = No distant metastases
M1 = Distant metastases
M1a = Lung
M1b = Other distant sites, including lymph nodes
The AJCC stage grouping is provided in the AJCC Cancer Staging Manual, 7th edition.
The Musculoskeletal Tumor Society staging system stages sarcomas according to grade and compartmental localization. The Roman numeral reflects the tumor grade.
Stage I: Low grade
Stage II: High grade
Stage III: Any-grade tumor with distant metastasis
Stage A: Confined to bone
Stage B: Extending into adjacent soft tissue
Stage IA tumor is a low-grade tumor confined to bone, and a stage IB tumor is a low-grade tumor extending into soft tissue, and so forth.
Because the histologic subtypes of sarcoma require specific treatment, the following sections will focus on the general principles of the treatment of each of these tumor types.
V. OSTEOSARCOMA
High-grade OS, also called conventional osteosarcoma, is a tumor with a poor prognosis in the absence of effective chemotherapy. Two randomized clinical trials comparing the administration of neoadjuvant and/or adjuvant chemotherapy have led to the conclusion that the administration of cytotoxic chemotherapy reduces the risk of metastatic relapse, which is the primary cause of death in these patients.3,4,5 Conventional osteosarcoma should be distinguished from low-grade juxtacortical or parosteal OS, which is rarer and often affects the lower femur in young women, and should not receive cytotoxic chemotherapy unless a dedifferentiated component exists in the tumor. OS is the most common primary bone sarcoma, typically affecting patients 10 to 25 years old, in two-thirds of cases in the lower femur or upper tibia. About 10% of patients have metastases at initial diagnosis. Relapses of OS occur most often in the lung, more rarely in the bones. Because conventional osteosarcoma is a high-grade tumor, by definition, and is accompanied by a soft tissue mass in 90% or more of patients, it is usually staged as IIB or IIIB, depending on the demonstration of metastatic disease in lung or bone.
A. Treatment strategy
The management of high-grade OS is multimodal, generally requiring the administration of specific cytotoxic chemotherapy regimens in a neoadjuvant setting, followed generally after 3 months of neoadjuvant treatment by a surgical resection of the primary tumor. The administration of neoadjuvant chemotherapy makes it possible to organize conservative surgery, in particular, the elaboration of the specific prosthesis for the replacement of the affected joint.5,6,7,8 The rules of surgical resection of the tumor are those of all sarcomas, with an en bloc complete macroscopic resection aiming for R0 staging (no tumor cells on the margins of the resection specimen). Adjuvant chemotherapy is administered after the local treatment and is usually guided by and adapted to the quality of response observed on the resection specimen after neoadjuvant treatment.1,2,9 This parameter, the percentage of residual tumor cells in the tumor, is one of the most important prognostic factors in high-grade OS. When surgical removal of the tumor is not possible, or when it is exceedingly mutilating, exclusive radiotherapy is generally discussed, even though the local control rate remains poor using radiotherapy alone in this histologic subtype (in contrast to ES). When the tumor has been treated by a primary excision of the whole tumor, adjuvant chemotherapy is proposed, with a discussion of a surgical removal of the tumor bed at the multidisciplinary board.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


