Therapy
Mechanism of action
Side effects
LH-RH agonists (buserelin, goserelin, leuprolide, and triptorelin)
Downregulation of LH-RH receptor
Flushes, weight increase, impotence, loss of libido, fatigue, decreased muscle mass, osteoporosis, anemia, metabolic syndrome, cardiovascular disorders, cognitive disorders, and “testosterone flare”
LH-RH antagonists (degarelix)
Competitive occupancy of LH-RH receptor
The same side effects of agonists with a putative lower incidence of cardiovascular, joint, musculoskeletal, and urinary tract events; no “testosterone flare”
Nonsteroidal antiandrogens (bicalutamide, flutamide, and nilutamide)
Competitive occupancy of AR
Anemia, liver toxicities, gynecomastia, mastalgia, and gastrointestinal disorders
Steroidal antiandrogens (cyproterone acetate)
Competitive occupancy of AR and gonadotropin release blockade
Cardiovascular events, gynecomastia, mastalgia, loss of libido, and sexual impotence
Abiraterone acetate
Inhibition of residual and adrenal androgen synthesis
Hypokalemia, hypertension, and fluid overload
Orteronel
Reversible inhibition of residual and adrenal androgen synthesis
Nausea, vomiting, fatigue, hypokalemia, and hypertension
Enzalutamide
Competitive occupancy of AR with high affinity, inhibition of AR nuclear translocation, and interaction with DNA
Cephalea, fatigue, hypertension, flushes, and seizures
10.2.1 Inhibitors of Gonadal Androgen Synthesis
As already mentioned, the surgical ablation of the gonads is the most effective and cheap way to obtain androgen deprivation and doesn’t expose the patient to any risk of paradoxical flare of the disease [18]. Medical castration is achieved by interfering with the release of gonadotropins at the pituitary level, and this treatment is considered an acceptable alternative to surgical castration, which is better tolerated by patients from a psychological point of view due to its putative reversibility [18–20]. Medical androgen deprivation has been accomplished through estrogens (diethylstilbestrol), luteinizing hormone-releasing hormone (LH-RH) agonists, and LH-RH antagonists. However, severe thromboembolic and cardiac side effects have limited the use of estrogenic therapy over time [20].
LH-RH (also known as gonadotropin-releasing hormone or GnRH) is secreted in the hypothalamic area of the brain in a pulsatile fashion, with pulses occurring every 60–90 min. The binding of LH-RH to specific plasma membrane receptors on pituitary gonadotrope cells is the step necessary to activate the synthesis and release of LH and FSH. Once LH enters the general circulation, it acts on the Leydig cells in the testes to control testosterone synthesis [21]. LH-RH agonists (buserelin, goserelin, leuprolide, and triptorelin) override this pulsatile control of the pituitary by providing continuous stimulation, which eventually leads to the downregulation of pituitary LH-RH receptors, the consequent reduction in LH and FSH release, and, therefore, the consequent suppression of testosterone levels [22] (Fig. 10.2). The chronic administration of LH-RH agonists achieves a deep and long-standing serum testosterone fall [19]; however, at the beginning of treatment, the physiological spiking of LH levels with consequent testosterone surge can determinate a transient disease flare, sometimes causing patients clinical worsening due to pain increase, spinal cord compression, or urethral obstruction [23, 24]. For this reason, the concurrent administration of an antiandrogen at the beginning of treatment with LH-RH agonists is usually recommended, especially in patients with bone metastases who have a higher risk of clinical complications from the flare caused by the early surge of testosterone [25, 26]. LH-RH antagonists (abarelix, degarelix) bind directly to LH-RH receptor at the pituitary cell level competing with native LH-RH, thus inducing an immediate suppression of LH, FSH, and testosterone circulating levels. Therefore, they are not associated with the initial surge of testosterone observed after agonist administration, and for this reason they don’t require the concurrent administration of an antiandrogen [27]. Two randomized trials evaluated the efficacy of the LH-RH antagonist abarelix depot in suppressing testosterone levels compared to leuprolide or leuprolide plus an antiandrogen. Abarelix not only achieved a comparable suppression of testosterone levels in the range of castrated levels but also achieved a more prompt reduction in the serum levels of this hormone without any flare effect [27, 28]. However, in 2009, this drug was withdrawn by the US FDA because of the occurrence of severe systemic allergic reactions subsequent to its administration. Allergic reactions are very uncommon with degarelix, a second-generation LH-RH antagonist. Therefore, this compound was approved by the US FDA and EMA for the treatment of prostate cancer as an alternative to LH-RH agonists [28]. The approval was granted based on the results of a phase III study which demonstrated the non-inferiority of degarelix compared with leuprolide in maintaining castration levels of testosterone (i.e., ≤0.5 ng/ml) for 1 year of treatment (primary endpoint of the study) in patients candidates to ADT [29].
Adverse events generally related to ADT include flushes, weight increase, impotence, loss of libido, fatigue, decreased muscle mass, osteoporosis, anemia, metabolic syndrome, and cardiovascular disorders [30–32]. Recent evidences also suggest the possible association between ADT and the appearance of cognitive disorders [33, 34]. A recent meta-analysis of five randomized trials comparing antagonists to agonists provides initial evidence about the putative superiority of antagonists on OS as well as on the lower incidence of ischemic cardiovascular events, particularly in patients with pre-existing cardiovascular disorders [35]. Noteworthy, the different incidence of cardiovascular events among patients allocated to degarelix was more evident during the first 6 months of treatment [30]. It is still unclear why the “protective effect” of LH-RH antagonists comes out especially in the first months of treatment. It was assumed that it might be mediated by the effect of treatment on the early detachment of arterial plaques, which, in turn, is probably mediated by the different behavior of FSH during treatment with agonists or antagonists or by immunologic mechanisms involving IL-2 [36, 37]. The previously mentioned meta-analysis also showed a reduced incidence of joint, musculoskeletal, and urinary tract adverse events in favor of LH-RH antagonists [35]. Noteworthy, LH-RH antagonists do not appear to be cross-resistant with LH-RH agonists, as shown in the retrospective study by Crawford et al., who demonstrated that patients initially progressed during treatment with an LH-RH agonist could be rescued after switching them to degarelix [23]. More recently, the results achieved with TAK 385, an oral LH-RH antagonist, confirmed the ability of this compound in achieving a prompt and durable suppression of testosterone levels, almost comparable to that induced by LH-RH agonist, with the advantage of being devoid of the local effects at the injection site that occur in about 40 % of the patients treated with degarelix [38]. Large prospective trials, specifically designed to compare the toxicity profile and survival outcomes of LH-RH agonists and antagonists, are still missing, and up to now the preference for antagonists is supported in selected patients, namely, those with cardiovascular comorbidities or at higher risk of tumor flare.
10.2.2 Inhibitors of Adrenal Androgen Synthesis
Abiraterone acetate was developed to interfere with residual androgen synthesis in castrated patients in a more selective and safer manner compared to the older adrenal steroidogenesis inhibitors, such as ketoconazole [39, 40]. In fact, this novel steroidal antiandrogen selectively inhibits the cytochrome P450 17α-hydroxylase/17,20-lyase (CYP17) activities. CYP17 is located in the endoplasmic reticulum of the testes and of the adrenals [41] and catalyzes two sequential reactions leading to the conversion of pregnenolone and progesterone into their 17α-hydroxy-derivatives and, subsequently, to dehydroepiandrosterone (DHEA) and androstenedione (ASD) [40]. ASD and DHEA are converted to testosterone through 17-beta-hydroxy-steroid dehydrogenase activity; testosterone is, in turn, converted to dihydrotestosterone (DHT), the biologically active compound, by 5α reductase. The inhibition of CYP17 thus decreases circulating levels of DHEA, ASD, testosterone, and DHT (Fig. 10.2). However, the blockade of abovementioned enzymatic pathways leads to the suppression of the negative adrenocorticotropic feedback, which, in turn, results in an exceeding production of precursor steroids with high mineralocorticoid activity. This effect is responsible for the most frequent adverse events observed during abiraterone treatment, namely, hypokalemia, hypertension, and fluid overload. The concomitant use of low-dose glucocorticoids, which block the adrenocorticotropic feedback, is therefore required to decrease the frequency and severity of these events [42, 43]. Initial phase I dose-escalation studies with abiraterone showed a strict relationship between the degree of testosterone suppression and the percentage of objective tumor responses [44, 45]. Indeed, a significant inhibition in testosterone levels was demonstrated in both castrate and non-castrate patients; however, a reactive LH rise was observed in non-castrate males, suggesting the opportunity of maintaining gonadal suppression during treatment with abiraterone [45]. Li et al. have recently demonstrated that abiraterone is converted to a more active metabolite, the delta-4-abiraterone (D4A). Beyond blocking the enzymatic pathways required to DHT synthesis through an increased affinity for CYP17, this metabolite directly competes for AR transcription, through a mechanism similar to enzalutamide. These findings provide an additional explanation for abiraterone’s activity in prostate cancer [46]. Moreover, interfering with abiraterone pharmacodynamics by adding an inhibitor of 5-alpha-reductase, which induces an increase in D4A levels, might increase treatment efficacy and overcome the resistance to this drug [46]. The use of dexamethasone in place of prednisone might also overcome the resistance to abiraterone, due to the higher affinity of dexamethasone for the glucocorticoid receptors which might be also involved in abiraterone resistance [47, 48]. Orteronel (TAK-700) is another inhibitor of adrenal androgen synthesis. Differently from abiraterone, this is a nonsteroidal, reversible inhibitor of CYP17. Results of a phase II trial preliminarily showed that orteronel (with or without prednisone) greatly decreased the plasma concentrations of testosterone, DHEA-S, and prostate-specific antigen (PSA) in patients with metastatic CRPC (mCRPC) [49]. However, two subsequent phase III trials in patients previously treated with docetaxel or chemo-naïve (ELM-PC 5 and ELM-PC 4 trials) failed to confirm the ability of orteronel to impact on patients’ survival (as it was the case for abiraterone; see next paragraphs); therefore, this drug did not obtain the US FDA approval [50, 51].
10.2.3 First-Generation Antiandrogens
Differently from therapies which act by interfering with the hypothalamic–pituitary–gonadal axis, antiandrogens compete directly with circulating androgens for binding sites on their receptors within the prostate cells, thus promoting apoptosis and inhibiting prostate cancer growth [52, 53] (Fig. 10.1). These effects can be achieved either by steroidal compounds like cyproterone acetate or by nonsteroidal moieties (bicalutamide, flutamide, nilutamide) [54]. Differently from nonsteroidal antiandrogens, cyproterone acetate also blocks gonadotropin release at the pituitary level (depending on the dose employed) causing a decrease in testosterone plasma levels too, leading to a sort of maximal androgen blockade [55, 56]. However, cardiovascular adverse events have been observed in more than 20 % of patients treated with this progestin [37]. These relevant side effects, which also include liver toxicity and fulminant hepatitis, have progressively narrowed the indications to the use of cyproterone acetate in prostate cancer [57, 58]. In contrast to cyproterone acetate, which causes loss of libido and sexual impotence, nonsteroidal antiandrogens have been shown to preserve sexual function if used as monotherapy. These drugs are also associated with a better physical capacity and quality of life; moreover, the treatment with these compounds has favorable effects on hemoglobin levels and bone mineral density [52, 59–61]. However, mild- to moderate-grade gynecomastia and mastalgia occur virtually in all patients, due to the increase in circulating estrogen levels which is the consequence of antiandrogen monotherapy [62]. This mechanism explains the efficacy of tamoxifen in preventing or in managing gynecomastia and breast pain in these patients [63]. Abnormal liver function tests have been observed with all nonsteroidal therapies [52]. Serious hepatotoxicity was rarely observed with bicalutamide and nilutamide, but a few cases of fulminant liver failure have been described with flutamide [62, 64]. Nilutamide has been associated with a higher incidence of adverse effects than the other nonsteroidal antiandrogens, including some peculiar toxicities like interstitial pneumonitis, delayed adaptation to darkness, and alcohol intolerance [62, 65]. Although no direct comparisons are available, bicalutamide appears to be the first-generation antiandrogen with the most favorable safety and tolerability profile [52]. As it will be discussed in the next paragraphs, these compounds have been developed to be used in combination with ADT, to prevent tumor flare or to improve treatment efficacy in the frame of a total androgen-blockade strategy [66]. However, they have been proven to be also effective as monotherapy, but only in selected groups of nonmetastatic patients. There is no indication that they might replace ADT in patients with metastatic disease, namely, those with bone metastases.
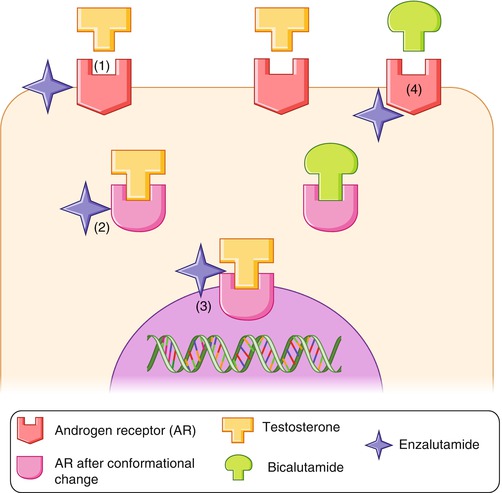
Fig. 10.1
Mechanisms of action of bicalutamide and enzalutamide. Bicalutamide competes directly with circulating androgens for binding sites on their receptors within the prostate cells. Enzalutamide inhibits prostate cancer cells acting at different levels: (1) It inhibits AR–testosterone binding with higher affinity than bicalutamide. (2) Receptor inhibition blocks the activational change induced by AR–testosterone binding. (3) It inhibits AR–testosterone nuclear translocation and DNA transcription. (4) Enzalutamide lacks partial AR agonist activity that occurs with bicalutamide resistance (From: Hoffman-Censits and Kelly [103])
10.2.4 Second-Generation Antiandrogens
First-generation antiandrogens, such as bicalutamide or flutamide, have modest efficacy in the setting of AR overexpression or of specific mutations in the AR ligand-binding domain [67]. New antiandrogens, like enzalutamide, were developed to overcome these limits. Enzalutamide has fourfold greater binding affinity for AR than bicalutamide; it reduces the efficiency of AR nuclear translocation and impairs both DNA binding to androgen response elements and recruitment of co-activators [54, 68, 69] (Figs. 10.1 and 10.2). Enzalutamide is also active against prostate cancer cell lines bearing the W741C AR point mutation that is known to confer resistance to bicalutamide. The superiority of enzalutamide over bicalutamide was recently demonstrated by two randomized controlled trials: the TERRAIN trial, which compared these two drugs in combinations with ADT in mCRPC [70], and the STRIVE trial, which compared enzalutamide versus bicalutamide in nonmetastatic or metastatic CRPC [71]. In the TERRAIN trial, a double-blind phase II study, 375 patients were randomly assigned to enzalutamide (184 patients) or bicalutamide (191 patients). Castrated levels of testosterone were maintained in both arms. Patients in the enzalutamide group had significantly improved median progression-free survival (PFS) duration (15.7 months) compared to patients in the bicalutamide group (5.8 months; HR: 0.44; p < 0.0001). In the STRIVE trial, patients with bone metastases represented about 55 % of the whole population on study. In this trial too, castrated levels of testosterone were required. Again enzalutamide was associated with a significant improvement in all study endpoints, including median time to PSA progression (HR: 0.19, p < 0.001) and PFS (median PFS duration: 16.5 vs. 5.5 months HR: 0.24, p < 0.001). The side effects reported in this study were comparable between groups; in fact, grade ≥3 adverse events and treatment-related deaths occurred in 36 % of patients and in 3 % of patients in each group, respectively. This study confirmed fatigue and hypertension being the most common clinically relevant adverse events. Seizures were not reported in this study but were previously observed in pivotal trials. Seizures are dose dependent and are thought to be related to the inhibition of gamma-aminobutyric acid (GABA) receptors in the brain [72]. Although the incidence of seizures in pivotal trials was small, it should be taken into account that patients eligible to these studies were required to have a low seizure risk at the time of enrolment (no prior seizures, no brain metastases, no recent stroke, no concomitant medications known to lower the seizure threshold), suggesting that the true incidence of seizures in an unselected patient population might be higher. Therefore, the administration of enzalutamide must be carefully evaluated in patients with a medical history of seizures or predisposing conditions.
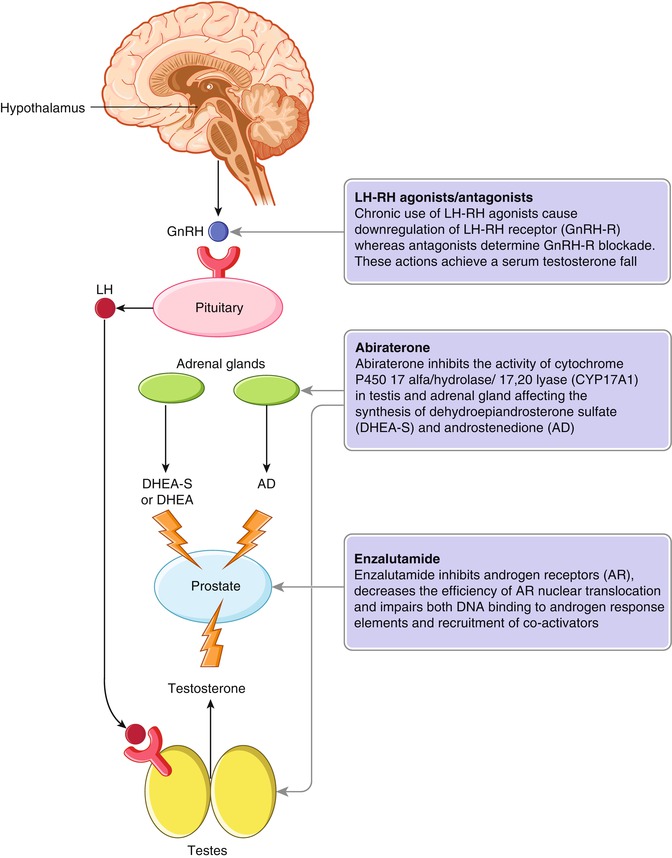




Fig. 10.2
Mechanisms of action of LH-RH agonists/antagonists, enzalutamide, and abiraterone. Chronic use of LH-RH agonists leads to downregulation of the LH-RH receptor (GnRH‑R), whereas antagonists provide immediate GnRH‑R blockade, achieving a deep and long-standing serum testosterone fall (a). Abiraterone selectively inhibits the cytochrome P450 17α-hydroxylase/17,20-lyase (CYP17A1) activities, located in the endoplasmic reticulum of testes and adrenals (b). Enzalutamide inhibits androgen receptor (AR) with high affinity, reduces the efficiency of AR nuclear translocation, and impairs both DNA binding to androgen response elements and recruitment of co-activators. DHEA dehydroepiandrosterone, GnRH gonadotropin-releasing hormone, LH luteinizing hormone, DHEA–S dehydroepiandrosterone sulfate, AD androstenedione, II (From: Watson et al. [104])
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
