C2. Bone marrow failure
Bone marrow failure has been divided into four groups:
• Aplastic anaemia
• Disorders characterised by a pancytopenia
• Disorders characterised by a single cytopenia
• Disorders characterised by a leucoerythroblastic blood picture.
Aplastic anaemia
Aplastic anaemia is a prime example of bone marrow failure. It is characterised by reduced bone marrow production of all three cell lineages: erythrocytes, granulocytes and platelets. This reduction causes peripheral blood pancytopenia and may be either acquired or inherited. In severe aplastic anaemia, the granulocyte or neutrophil count is <0.5 × 109/L, the platelet count is <20 × 109/L and the reticulocyte count is <1%. The anaemia may be normocytic or macrocytic. The bone marrow is hypocellular, with empty fragments and increased fat spaces.
Aplastic anaemia may be inherited or acquired. Acquired aplastic anaemia may be secondary to certain drugs, chemicals and viruses (parvovirus, Epstein-Barr virus, cytomegalovirus and human immunodeficiency virus). Inherited aplastic anaemia includes disorders such as Fanconi anaemia, dyskeratosis congenita and Shwachman-Diamond syndrome. Refer to Fig C2-1.
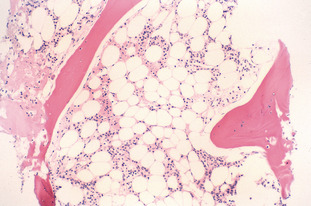
Pancytopenias
Fanconi anaemia (FA)
FA is an autosomal-recessive disorder encompassing a group of patients with familial aplastic anaemia and physical anomalies. There is clinical variability amongst patients with FA. Three groups of patients have been described: those with physical anomalies and normal haematological findings, those with no physical anomaly but with abnormal haematological findings and those with both a physical anomaly and abnormal haematological findings.
The haematological features in Fanconi anaemia lead to the gradual onset of bone marrow failure. Patients present initially with thrombocytopenia that is followed by neutropenia and then anaemia. The red cells are often macrocytic with an MCV of more than 100 fL. There is an increase in HbF production. Erythropoietin levels are also increased. The bone marrow is hypercellular in early FA (before the development of pancytopenia). When the patient becomes aplastic, the bone marrow is hypocellular and fatty with few haemopoietic elements. There is an increase in the number of lymphocytes, reticulum cells, mast cells and plasma cells. FA is characterised by abnormal chromosome fragility. Spontaneous breaks, gaps, rearrangements, exchanges and endoreduplications occur. This abnormal chromosome fragility can be exacerbated by the addition of diepoxybutane (DEB) in chromosome culture. FA homozygotes have a mean of 8.96 breaks per cell with DEB culture compared with 0.06 breaks in normal subjects.
Dyskeratosis congenita (DC)
DC is a disorder of the mucocutaneous and haemopoietic systems. The mucocutaneous aspect of the disorder is characterised by a triad of events: pigmentation of the upper body, mucosal leucoplakia and nail dystrophy. The haemopoietic aspect of the disorder is characterised by aplastic anaemia occurring in the second decade of life and in approximately 50% of cases. A small percentage of cases have a predisposition to develop cancer in the third to fourth decade of life.
DC has three patterns of inheritance: X-linked recessive, autosomal-recessive and autosomal-dominant.
The haematological features of DC are anaemia, leucopenia and thrombocytopenia. The bone marrow is initially hypercellular but gradually becomes hypocellular, culminating in aplastic anaemia. The red cells are macrocytic and HbF is increased. Chromosome fragility in DC is variable when DEB is added to culture. Refer to Fig C2-2.
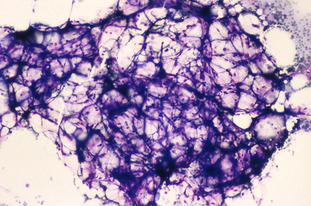
Shwachman-Diamond syndrome (SDS)
SDS is an autosomal-recessive disorder characterised by pancreatic dysfunction, malabsorption, steatorrhoea, failure to thrive, short stature and (in some cases) mental retardation. Fatty infiltration of the pancreas leads to pancreatic insufficiency with low or absent duodenal trypsin, amylase and lipase.
The main haematological feature of SDS is neutropenia. Anaemia and thrombocytopenia may occur in some cases. The neutropenia is intermittent rather than constant and is associated with skin infections and pneumonia. Abnormal neutrophil mobility, migration and chemotaxis lead to recurrent infections. An increase in HbF, even without anaemia, is common. Bone marrow hypoplasia with maturation arrest in the myeloid line is present.
Chromosomes are normal and there is no increased breakage following clastogenic stress with DEB.
Single cytopenias
Diamond-Blackfan anaemia (DBA)
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


