Fig. 3.1
Bone metastasis of high-grade prostate cancer
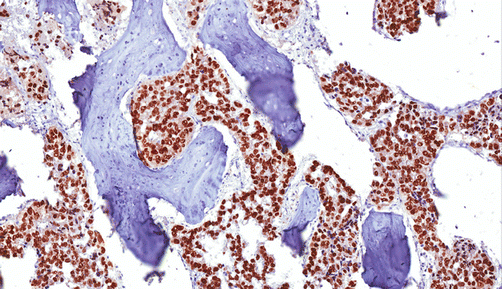
Fig. 3.2
Bone metastasis of prostate cancer. The neoplastic cells are immunostained with antibody against prostein
Understanding the mechanisms by which PCa cells migrate into the bone will represent a major step forward for the early detection of bone involvement in patients with PCa and for the design of agents specifically targeting bone homing and preventing this phenomenon.
In this chapter, we describe the main mechanisms involved in PCa bone homing and metastasis.
3.2 Homing of PCa Cells to the Bone: The Role of Cellular Plasticity
The biological scenario underlying bone homing has not been completely clarified and includes a huge variety of mechanisms. Among them, the selective pressure exerted by bone microenvironment seems to majorly contribute to tumor cell homing [11, 12]. Indeed, it has been shown that the frequency of mitochondrial DNA mutations is significantly higher in BMs compared to both soft tissue metastases and the primary tumor [11]. Otherwise, the contribution of osteoclasts does not seem to be relevant in the initiation phase of PCa bone metastasization [12].
Cellular plasticity is the ability of differentiated cells of deviating to other cell types when exposed to different conditions. Through this phenomenon, differentiated cells can acquire cancer stem cell (CSC) properties under specific oncogenic insults, leading to aberrant cell reprogramming and, as a consequence, to a series of diseases including cancer [13]. Indeed, CSC status and EMT and mesenchymal-epithelial transition (MET) are interconnected reversible dynamic processes that facilitate cells to adapt to stimuli from altered microenvironments [14]. In breast cancer, CSCs have been shown to reversibly switch from mesenchymal-like to epithelial-like states. Bone colonization from breast CSCs can induce the co-expression of mesenchymal and epithelial markers and the transition from a CD44+/CD24− to a CD44−/CD24+ phenotype [15, 16], thus underlining the complexity of tumor cell bone homing and stem cell trafficking.
At present, data on the role of cellular plasticity in bone homing and development of BMs from PCa are still inconclusive [17]. Thus, while several studies have observed that the interaction between androgen refractory PCa cell lines and bone stromal cells led to the acquisition of a mesenchymal phenotype via EMT characterized by a switch from E- to N-cadherin expression [14, 18], the study published by Josson et al. has reported increased E-cadherin expression as a result of a similar interaction [19].
Cellular plasticity may be the result of physiological stimuli and induced by therapeutic agents. Hypoxia seems to majorly contribute to the promotion of cellular plasticity [20], and E-cadherin is implicated in the regulation of the response of cancer cells to hypoxia by inducing the expression of hypoxia-inducible factor-1α (HIF-1α) [21]. On the other hand, the selective pressure exerted by androgen-deprivation therapy (ADT) may give rise to CSCs as well as EMT and neuroendocrine (NE) differentiation, which are associated with PCa growth, metastasis, and resistance to therapies [22, 23].
Based on these data, several strategies are emerging to modulate the contribution of bone microenvironment in patients with PCa. Among them, abiraterone acetate seems to represent an effective strategy in this context. Food and Drug Administration (FDA) approved this drug in combination with prednisone on April 2011 for metastatic castration-resistant PCa following docetaxel and on December 2012 for metastatic castration-resistant PCa before chemotherapy. In 2015, Santini and his group have first revealed that abiraterone acetate can affect the differentiation and activity of osteoclasts by inhibiting marker genes including TRAP, cathepsin K, and metalloproteinase-9 (MMP-9) [24]. Moreover, abiraterone acetate can promote osteoblast differentiation and the deposition of bone matrix through the upregulation of specific genes such as ALP and osteocalcin [24]. However, the role of abiraterone acetate in modulating bone homing has not been clarified so far and seems to merit further investigations.
3.3 Epithelial-Mesenchymal Plasticity in Bone Homing
During embryonic morphogenesis, epithelial cells exhibit enormous plasticity and transit into a mesenchymal state by activating the EMT process. Through this program, epithelial cells lose their junctions and produce vimentin filaments, thus enhancing their ability to migrate and invade during developmental morphogenesis [25, 26] (Fig. 3.3). During EMT, E-cadherin gene transcriptional repression, promoter methylation, and protein phosphorylation and degradation have been observed [27, 28]. On the other hand, N-cadherin, fibronectin, and cell surface proteins CD44 [29] and integrin β6 [30].
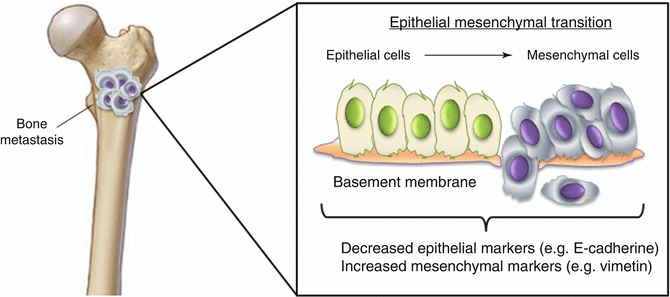
Fig. 3.3
Epithelial-mesenchymal transition (EMT) in prostate bone metastases (BMs)
The expression of EMT and its reversal MET are fundamental processes for the development and progression of genitourinary tumors [31–34]. In PCa, the functional cross talk between EMT and castration resistance, which is crucial during prostate carcinogenesis, is mediated by the Twist1/AR signaling axis and promoted by the activity of transforming growth factor-β (TGF-β) [35].
TGF-β upregulates a variety of genes, such as prostate transmembrane protein, androgen induced 1 (PMEPA1), that is a regulator of TGF-β and correlates with tumor aggressiveness and BMs [36]. Interestingly, the knockdown of PMEPA1 has been associated with increased prometastatic gene expression and bone homing in PCa mouse models [36].
Furthermore, TGF-β expression resulted increased in BMs compared to visceral PCa metastases in a series of 149 visceral and BMs from 62 patients with castration-resistant PCa [37]. In this study, nuclear Twist, Slug, and Zeb1 localization and an EMT-like phenotype were present only in a small subset of castration-resistant PCa BMs [37].
E-cadherin is involved in maintaining the pluripotent and self-renewal ability of prostate CSCs [28, 38, 39]. Interestingly, E-cadherin loss, which is crucial for the acquisition of PCa migratory properties and bone homing, is associated with activated androgen receptor (AR) [40] and correlates with PCa progression and Gleason score [41], while increased N-cadherin has been reported to predict clinical recurrence in PCa patients following RP [42], thus representing a potential therapeutic target in castration-resistant PCa [43].
3.4 The Role of Chemokines and Their Receptors
Chemokines are a superfamily of low-molecular-weight proteins including inducers and inhibitors of angiogenesis. The altered balance between these stimuli can lead to chronic inflammatory diseases, as well as to tumor initiation and spreading [44].
The chemokine family includes CXC ligand 1 (CXCL1) to CXCL16. These ligands interact with the CXC chemokine receptors (CXCR1–CXCR5), members of the rhodopsin-like seven-transmembrane G protein-coupled receptor family, to exert their activity [45, 46].
Several studies demonstrated that chemokines and their receptors are implicated in chemotaxis of cancer cells toward bone and the lymph nodes. Among CXCRs, CXCR4 has been shown to play an essential role for both normal prostate tissue and PCa development and progression. CXCR4 binds to CXCL12/SDF-1, which is implicated in the maintenance of leukocyte trafficking during homeostasis. In PCa, SDF-1 supports the invasion of PCa cell lines through basement membranes, which is conversely inhibited by anti-CXCR4 antibodies [47].
It is interesting to note that PCa cell androgen-mediated motility seems to be dependent on functional CXCR4/CXCR7 heterodimers [48]. Moreover, prostate CSCs expressing CXCR4 compete with HSCs for bone marrow niches. Blocking CXCR4-dependent bone homing, the formation of BMs is suppressed [49]. It should be noted that the expression of CXCR4 is low in BMs, suggesting that this receptor may be essential for bone homing but not for distant tumor growth [50]. The potential role of CXCR4 as a potential therapeutic target is also sustained by the evidence that Plerixafor, a CXCR4 inhibitor, seems to be effective in PCa xenograft mouse models [51]. Furthermore, homing may be also prevented by agents such as pertussis toxin, which inhibits G proteins, and chelerythrine chloride, which inhibits protein kinase C.
3.5 The Role of MicroRNAs
Regarding the role played by microRNAs (miRNAs) in the control of PCa metastases, they are involved in regulating the complex metastatic cascade at multiple levels. Fu and his group have compared the expressions of four miRNAs (miR-335, miR-543, miR-196, and miR-19a) between primary PCa and BMs [52]. By using reverse transcription-quantitative polymerase chain reaction, they showed that the four miRNAs were significantly downregulated in BMs compared to PCa. Additionally, miR-335 and miR-543 downregulation was confirmed in 20 paired primary tumors and BMs [52]. Exogenous miR-335 and miR-543 significantly reduced the expression level of endothelial nitric oxide synthase (eNOS) and markedly affected the migratory and invasive properties of PCa cells in vitro [52].
It has been shown that loss of miR-15 and miR-16, together with increased miR-21 expression, promotes PCa metastasization and bone homing [53]. In the same view, dysregulation of the epidermal growth factor receptor (EGFR) signaling pathway seems to sustain PCa bone homing by constraining the tumor-suppressive role of miR-1 and promoting the oncogenic activation of Twist1 [54].
Related posts:
 Circulating Tumor Cells (CTCs) and Metastatic Prostate Cancer (mPCa)
Economic Impact of Prostate Cancer Bone Metastases
Circulating Tumor Cells (CTCs) and Metastatic Prostate Cancer (mPCa)
Economic Impact of Prostate Cancer Bone Metastases
 Bone Metastases from Prostate Cancer: Radiotherapy
Bone Metastases from Prostate Cancer: Radiotherapy
 Bone Metastases from Prostate Cancer: Hormonal Therapy
Bone Metastases from Prostate Cancer: Hormonal Therapy
 Imaging of Glycolysis with 18F-FDG PET
Imaging of Glycolysis with 18F-FDG PET
 Surgery: Treatment of Oligometastatic Disease
Surgery: Treatment of Oligometastatic Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


