Blood Transfusion
![]()
BLOOD CELL ANTIGENS
Red blood cell (RBC) antigens are classified according to their biochemical, phenotypic, and immunologic characteristics. Based on these characteristics, they have been separated into blood group systems. Currently 30 major blood group systems are recognized, the most clinically relevant being ABO, Rh, Kell, Kidd, and Duffy. Clinically important alloantibodies (specific for antigens not found on the individuals red cells) can cause destruction of transfused red cells or are implicated in hemolytic disease of the newborn (HDN).
The antiglobulin or Coombs test (see below) is used to detect antibodies to red cell antigens and in crossmatching compatible blood units for transfusion. When a clinically important alloantibody is present in a recipient’s serum, antigen-negative blood must be selected. If the alloantibody is against a very high frequency antigen (present in greater than 90% of individuals) or when multiple alloantibodies are present, procurement of compatible blood may be difficult or impossible. Occasionally, the presence of red cell autoantibodies in the recipient makes all units appear incompatible. Further investigations are necessary in these cases to rule out an underlying alloantibody.
“Naturally occurring” antibodies such as anti-A and/or anti-B are present in the absence of prior sensitizing stimulus, whereas development of most other alloantibodies requires prior sensitization by exposure to the corresponding red cell antigen in a previous transfusion or pregnancy.
![]() Blood group A individuals have naturally occurring anti-B
Blood group A individuals have naturally occurring anti-B
![]() Blood group B individuals have naturally occurring anti-A
Blood group B individuals have naturally occurring anti-A
![]() Blood group O individuals have naturally occurring anti-A and anti-B
Blood group O individuals have naturally occurring anti-A and anti-B
![]() Blood group AB individuals have neither anti-A nor anti-B
Blood group AB individuals have neither anti-A nor anti-B
![]()
LABORATORY DETERMINATION OF MAJOR BLOOD GROUPS
An individual’s blood group is determined by performing a forward and reverse grouping (ABO typing):
![]() In forward grouping, reagents of known antibody specificity (anti-A or anti-B) are added to the patient’s RBCs of unknown phenotype (A, B, AB, or O) and the mixtures are examined for visible agglutination; the absence of agglutination on combining the patient’s cells with anti-A or anti-B reagent indicates that the patient’s cells do not have the corresponding antigen. For example, red cells of group O will not agglutinate in the presence of anti-A and/or anti-B.
In forward grouping, reagents of known antibody specificity (anti-A or anti-B) are added to the patient’s RBCs of unknown phenotype (A, B, AB, or O) and the mixtures are examined for visible agglutination; the absence of agglutination on combining the patient’s cells with anti-A or anti-B reagent indicates that the patient’s cells do not have the corresponding antigen. For example, red cells of group O will not agglutinate in the presence of anti-A and/or anti-B.
![]() In reverse grouping, the patient’s serum is added to reagent cells of known phenotype (A, B, or O) and the mixtures are examined for visible agglutination; the presence of agglutination on combining the patient’s serum with reagent cells of A or B phenotype, indicates that the patient’s serum contains the corresponding antibody. For example serum containing anti-A and anti-B (blood group O) will agglutinate in the presence of both group A and group B red cells.
In reverse grouping, the patient’s serum is added to reagent cells of known phenotype (A, B, or O) and the mixtures are examined for visible agglutination; the presence of agglutination on combining the patient’s serum with reagent cells of A or B phenotype, indicates that the patient’s serum contains the corresponding antibody. For example serum containing anti-A and anti-B (blood group O) will agglutinate in the presence of both group A and group B red cells.
The forward and reverse groups must be consistent. A specific blood group may not be assigned with certainty until an ABO discrepancy is resolved.
Some common causes of apparent ABO discrepancies are1 presence of A or B subgroups,2 missing/ weakly reacting antibodies (can occur with newborns, the elderly, or in hypogammaglobulinemic states) or weak expression/absence of expected antigens (can occur with lymphoproliferative disorders and post hematopoietic stem cell transplantation, HSCT),3 presence of unexpected or nonspecific antibodies such as cold reactive alloantibodies or autoantibodies,4 interfering substances such as Wharton jelly from the umbilical cord of a newborn, or5 hyperproteinemic states causing Rouleaux formation.
![]()
ANTIGLOBULIN TEST
The antiglobulin test uses antibodies with specificity for immunoglobulins or complement to detect the presence of antibody (or complement) on the RBC surface or in a patient’s serum.
The direct antiglobulin test (DAT), or the direct Coombs test, detects the presence of antibody or complement coating RBC and may be positive in a variety of settings, including those listed below:
![]() Hemolytic transfusion reactions
Hemolytic transfusion reactions
![]() HDN (usually strongly positive)
HDN (usually strongly positive)
![]() Autoimmune hemolytic anemias (AIHA)
Autoimmune hemolytic anemias (AIHA)
![]() With some pharmacologic agents (penicillins, cephalosporins)
With some pharmacologic agents (penicillins, cephalosporins)
![]() After administration of intravenous immunoglobulin (IVIG) or plasma (passively acquired)
After administration of intravenous immunoglobulin (IVIG) or plasma (passively acquired)
![]() Post marrow or organ transplant (passenger lymphocyte syndrome)
Post marrow or organ transplant (passenger lymphocyte syndrome)
![]() Autoimmune diseases
Autoimmune diseases
![]() Some normal individuals
Some normal individuals
A positive DAT does not necessarily indicate in vivo hemolysis or shortened RBC survival. Falsepositive results may occur when Rouleaux formation is mistaken for agglutination.
The indirect antiglobulin test (IAT), or indirect Coombs test, is used to screen for antibodies when looking for compatible blood for transfusion (“type and screen”) and in the serologic cross-match (patient serum and donor/ reagent red cells). The IAT detects antibody present in the serum, but not bound to the RBC. When the IAT is positive, the antibody must be identified and the corresponding antigen always avoided in transfusions. A negative IAT does not necessarily indicate absence of alloantibodies: the titer of antibody may be below the level of detection or the antibody might be directed against a low incidence antigen (present in less than 1% of individuals) not present on reagent testing cells. A negative IAT does not guarantee that blood is compatible, nor does a weak IAT indicate that hemolysis is likely to be mild. A positive IAT always requires further investigation.
In AIHA, in which the antibody may be present in the serum and on the RBC of the patient, both the DAT and IAT may be positive.
![]()
BLOOD COMPATIBILITY
In general, blood components that contain more than 2 mL of RBC must be compatible with the patient’s plasma. Particular attention is paid to Rh type because less than 1 mL of RBC, a volume found in most platelet concentrates, is sufficient to sensitize an Rh-negative patient.1 Plasma-containing components, including platelet preparations, should be ABO compatible with the patient’s RBC when possible to prevent passive immune hemolysis from antibodies in the plasma (Table 24.1).

The most basic practical application of blood group serology involves the selection of compatible blood. The absence or presence of blood cell antigens can have important biological and clinical implications. Compatible blood takes time to prepare.
![]() In an emergency, group O Rh-negative RBC may be released uncrossmatched with the consent of the requesting physician; testing will be completed after release. Group-specific red cells and an abbreviated crossmatch can be prepared in 15 minutes.
In an emergency, group O Rh-negative RBC may be released uncrossmatched with the consent of the requesting physician; testing will be completed after release. Group-specific red cells and an abbreviated crossmatch can be prepared in 15 minutes.
![]() Fully tested red cells can be prepared in 45 to 60 minutes; cryopreserved RBC and fresh-frozen plasma (FFP) may take longer.
Fully tested red cells can be prepared in 45 to 60 minutes; cryopreserved RBC and fresh-frozen plasma (FFP) may take longer.
In the setting of transfusion or pregnancy in the previous 3 months a pretransfusion sample cannot be more than 3 days old.6
![]()
ABO INCOMPATIBILITY IN TRANSPLANT SETTINGS
Optimal results in HSCT rely on human leukocyte antigen (HLA) compatibility, so that selection of the recipient-donor pair is determined by similarity of HLA potentially at the expense of ABO compatibility. Because HLA and ABO genes are inherited independently, some (20%–40%) of these transplants will be ABO incompatible.1
Whereas ABO incompatibility does not appear to impact graft failure, potential complications of mismatches include acute or delayed hemolysis and delayed RBC engraftment.2–5
Minor Incompatibility
![]() Donor’s serum contains antibodies against RBC antigens of the recipient (e.g., recipient blood group A, B, or AB, and donor blood group O)
Donor’s serum contains antibodies against RBC antigens of the recipient (e.g., recipient blood group A, B, or AB, and donor blood group O)
![]() Prior to infusion of the stem cell preparation, plasma containing anti-A and anti-B can be removed to prevent immediate postinfusion hemolysis of the recipient’s RBC (plasma reduction)
Prior to infusion of the stem cell preparation, plasma containing anti-A and anti-B can be removed to prevent immediate postinfusion hemolysis of the recipient’s RBC (plasma reduction)
![]() Of minor ABO-incompatible transplant recipients, 10% to 15% may experience abrupt onset of hemolysis 5 to 15 days posttransplant when immune-competent B lymphocytes in the graft mount a response against the recipient RBC antigens (passenger lymphocyte syndrome)
Of minor ABO-incompatible transplant recipients, 10% to 15% may experience abrupt onset of hemolysis 5 to 15 days posttransplant when immune-competent B lymphocytes in the graft mount a response against the recipient RBC antigens (passenger lymphocyte syndrome)
![]() Hemolysis may be severe or even fatal unless recognized promptly
Hemolysis may be severe or even fatal unless recognized promptly
Major Incompatibility
![]() Recipient’s serum contains antibodies against the RBC antigens of the donor (e.g., recipient group O and donor group A, B, or AB; recipient group A or B and donor group AB).
Recipient’s serum contains antibodies against the RBC antigens of the donor (e.g., recipient group O and donor group A, B, or AB; recipient group A or B and donor group AB).
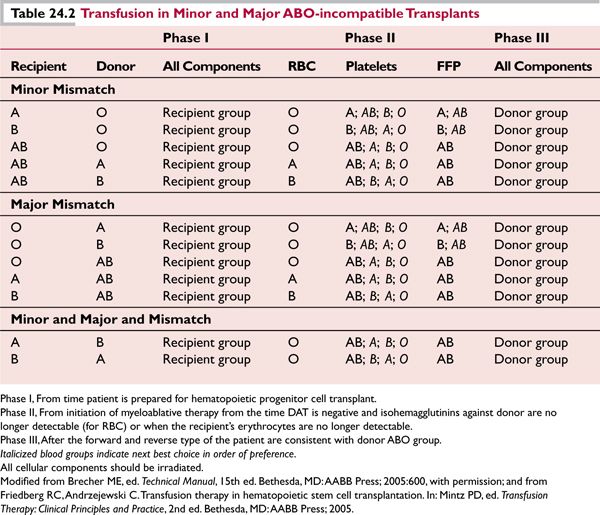
![]() Hemolysis of RBC in the stem cell preparation upon infusion may occur if the graft is not processed to remove RBC prior to infusion (red cell reduction).
Hemolysis of RBC in the stem cell preparation upon infusion may occur if the graft is not processed to remove RBC prior to infusion (red cell reduction).
![]() Post transplant, the recipient may produce antibodies against donor red cell antigens for months, especially with nonmyeloablative regimens.
Post transplant, the recipient may produce antibodies against donor red cell antigens for months, especially with nonmyeloablative regimens.
![]() RBC engraftment and erythropoiesis may be delayed, resulting in red cell aplasia.5,6
RBC engraftment and erythropoiesis may be delayed, resulting in red cell aplasia.5,6
Minor and major (bidirectional) incompatibility between the donor and recipient occurs when each has antibodies against the ABO blood group antigens of the other (combination of group A donor and B recipient, or vice versa).
Table 24.2 describes appropriate transfusion management during transplantation. All transfusion components must be irradiated.
Rh Incompatibility
Rh incompatibility occurs in 10% to 15% of stem cell transplants. Transfusion practice is analogous to that of major and minor ABO incompatibility, but the consequences are less severe.
For Rh-negative recipients of Rh-positive hematopoietic stem cell preparations
![]() Transplant product should be red cell reduced to decrease risk of alloimmunization similar to major ABO incompatibility
Transplant product should be red cell reduced to decrease risk of alloimmunization similar to major ABO incompatibility
For Rh-positive recipient from Rh-negative donor previously alloimmunized to the Rh antigen
![]() Monitor the patient for signs of delayed hemolysis (as in minor incompatibility).
Monitor the patient for signs of delayed hemolysis (as in minor incompatibility).
![]()
BLOOD COMPONENTS AND DERIVATIVES
Blood Components and Transfusion Therapy
Blood components can be separated from whole blood by centrifugation or by apheresis. Approximately 29 million blood components (RBCs, platelets, plasma, cryoprecipitate) are transfused annually in the United States.
Storage and infusion of blood products
![]() Blood components should be infused through standard 170 to 260 μm infusion filters to remove any clots that form during storage. 6
Blood components should be infused through standard 170 to 260 μm infusion filters to remove any clots that form during storage. 6
![]() An approved infusion pump may be used for strict control of transfusion rate. Nonapproved pumps may damage or hemolyze cells.
An approved infusion pump may be used for strict control of transfusion rate. Nonapproved pumps may damage or hemolyze cells.
![]() Bedside leukoreduction filters may be used when leukoreduction is indicated for whole blood, packed RBCs, and platelets that have not been leukocyte reduced prior to storage.
Bedside leukoreduction filters may be used when leukoreduction is indicated for whole blood, packed RBCs, and platelets that have not been leukocyte reduced prior to storage.
![]() Hypotensive reactions have been associated with bedside leukoreduction, especially in patients receiving angiotensin converting enzyme (ACE) inhibitors.
Hypotensive reactions have been associated with bedside leukoreduction, especially in patients receiving angiotensin converting enzyme (ACE) inhibitors.
![]() Allow blood to filter by gravity.
Allow blood to filter by gravity.
![]() Granulocyte concentrates must never be infused through leukoreduction filters.
Granulocyte concentrates must never be infused through leukoreduction filters.
![]() Whole blood and other cellular blood components may be infused with isotonic solutions: USP 0.9% NaCl (normal saline) and certain Food and Drug Administration (FDA) approved electrolyte solutions.
Whole blood and other cellular blood components may be infused with isotonic solutions: USP 0.9% NaCl (normal saline) and certain Food and Drug Administration (FDA) approved electrolyte solutions.
![]() Cellular blood products must never be infused with hypertonic or hypotonic solutions, for example, solutions containing glucose or calcium, such as D5W (5% dextrose in water) or lactated Ringer solution as hemolysis, clotting, or agglutination of RBC may result.
Cellular blood products must never be infused with hypertonic or hypotonic solutions, for example, solutions containing glucose or calcium, such as D5W (5% dextrose in water) or lactated Ringer solution as hemolysis, clotting, or agglutination of RBC may result.
![]() Medications should never be added to blood components.
Medications should never be added to blood components.
![]() Never store blood components in unmonitored refrigerators in nursing units or surgical suites; the risk for administration of blood components to the wrong patient increases in these cases.
Never store blood components in unmonitored refrigerators in nursing units or surgical suites; the risk for administration of blood components to the wrong patient increases in these cases.
![]() Return blood to storage (or blood bank) if transfusion is not started within 30 minutes of issue.
Return blood to storage (or blood bank) if transfusion is not started within 30 minutes of issue.
![]() Warming devices with internal monitors are available for blood products to avoid transfusion of large volumes of cold fluid.
Warming devices with internal monitors are available for blood products to avoid transfusion of large volumes of cold fluid.
![]() Blood components should never be warmed in uncertified devices (like microwave ovens or water baths) as hemolysis can result and can be lethal.
Blood components should never be warmed in uncertified devices (like microwave ovens or water baths) as hemolysis can result and can be lethal.
Most adverse transfusion reactions occur in the first 15 minutes:
![]() Administration of blood products should start slowly and under close observation.
Administration of blood products should start slowly and under close observation.
![]() The time of transfusion should not exceed 4 hours as the risk of bacterial growth increases with time at room temperature.
The time of transfusion should not exceed 4 hours as the risk of bacterial growth increases with time at room temperature.
![]() If transfusion is anticipated to take longer, the transfusion service can divide the unit into smaller aliquots.
If transfusion is anticipated to take longer, the transfusion service can divide the unit into smaller aliquots.
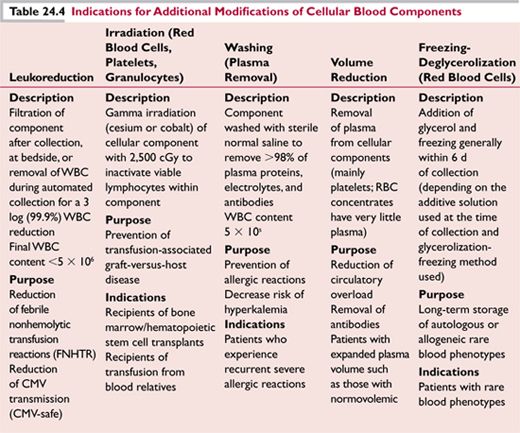
See Table 24.3 for blood component administration.
See Table 24.4 for indications for additional modifications to blood components.
Storage conditions for different blood components vary and are designed to maximize preservation and effectiveness:
![]() Red cells are refrigerated (at 1–6°C) for up to 42 days.
Red cells are refrigerated (at 1–6°C) for up to 42 days.
![]() Platelets are stored at room temperature and expire in 5 days.
Platelets are stored at room temperature and expire in 5 days.
![]() Plasma components are stored frozen for a year (at −18°C) or more (at −65°C), but must be thawed before use and therefore are not immediately available.
Plasma components are stored frozen for a year (at −18°C) or more (at −65°C), but must be thawed before use and therefore are not immediately available.
As with any medical treatment, blood transfusion requires informed consent:
![]() Patients must be advised of the indications and common adverse events as well as any potential alternatives to allogeneic transfusion.
Patients must be advised of the indications and common adverse events as well as any potential alternatives to allogeneic transfusion.
Whole Blood
A unit of whole blood typically has a volume of 450 to 500 mL and a hematocrit of 35% to 45%. Whole blood is rarely available and infrequently used.


Indications: Acute hypovolemia with red cell loss, massive transfusion, and exchange transfusion Not indicated: Chronic anemia (in which blood volume is often increased)
Whole blood is not a source of functional platelets or granulocytes, which deteriorate in less than 24 hours at refrigerator temperatures.
Red Blood Cells
RBC (packed red cells) are separated from whole blood by centrifugation. A unit of RBC contains approximately 200 mL and a hematocrit of 60% to 80%. In general, 1 unit of packed RBC will increase the hemoglobin (Hb) by 1 g/dL in an average-sized adult. In the average pediatric patient, transfusion of 8 to 10 mL/kg of RBC is expected to increase hemoglobin by 3 g/dL. The decision to transfuse should be based on assessment of symptoms, coexisting or underlying medical conditions, and the cause of the anemia and patients should not be transfused based solely on their Hb level. The single adequately powered prospective study (ICU patients) and numerous observational studies indicated that patients with cardiovascular disease are more sensitive to anemia and do better at a higher Hb level.7,8
Indications: Treatment of symptomatic anemia.
While it is generally accepted that patients with Hb <6 g/dL should be transfused and that transfusion is rarely required when it exceeds 10 g/dL, the interval between these values is an area of controversy. Practice guidelines support an Hb level of less than 7 g/dL as generally acceptable for the initiation of RBC transfusion in asymptomatic patients.8,9 Patients at particular risk for bleeding (thrombocytopenia, recent hemorrhage) should be maintained at a higher Hb.
Not indicated: RBC should not be transfused for volume expansion or nutritional purposes Transfusion is rarely indicated in otherwise treatable anemia, including anemia associated with Vitamin B12, iron, or folate deficiency; if symptoms are severe, these patients may benefit from a single-unit transfusion as the underlying cause is corrected.
Platelets
Platelets may be separated from whole blood shortly after collection (“random donor” or “whole blood-derived” platelet concentrates) or collected by apheresis (“single donor” or “apheresis platelets”). A therapeutic dose of platelets for an adult is 1 unit of platelets (5.5 × 1010 platelets) per 10 kg of body weight, which should increase the platelet count in an average-sized adult by approximately 5,000/μL. Each apheresis (single-donor) platelet product is expected to contain approximately 3 × 1011 platelets, roughly equivalent to 4 to 6 units of random donor platelets. Indications for use are the same for both preparations. Alloimmunized refractory patients may require single-donor HLA-matched platelets. Single-donor platelets also offer the additional advantage of decreased donor exposure and a lower risk of bacterial sepsis.
Indications: Prevention and treatment of hemorrhage in patients with thrombocytopenia or platelet function defects.
Not indicated: Bleeding unassociated with thrombocytopenia (in the absence of a clinically significant platelet function defect), other defects in hemostasis (such as factor deficiencies).
Platelets are usually contraindicated in thrombotic thrombocytopenic purpura (TTP),10 because they could potentially precipitate thrombosis, but this has been recently challenged.11 However, patients with TTP who develop life-threatening hemorrhage may benefit from a cautious trial of platelets.
The threshold for prophylactic platelet transfusion varies based on the patient’s underlying condition and likelihood of hemorrhage:
![]() A threshold of 10,000/μL is effective in preventing morbidity and mortality from bleeding in stable oncology patients undergoing chemotherapy.
A threshold of 10,000/μL is effective in preventing morbidity and mortality from bleeding in stable oncology patients undergoing chemotherapy.
![]() A platelet count greater than 50,000/μL is desirable prior to invasive procedures and in the immediate postprocedure period.
A platelet count greater than 50,000/μL is desirable prior to invasive procedures and in the immediate postprocedure period.
![]() Platelet counts closer to 100,000/μL may be prudent for patients at high risk for intracranial hemorrhage, such as those with cerebral leukostasis, or when undergoing neurosurgical or ocular procedures.
Platelet counts closer to 100,000/μL may be prudent for patients at high risk for intracranial hemorrhage, such as those with cerebral leukostasis, or when undergoing neurosurgical or ocular procedures.
![]() Stable chronically thrombocytopenic patients, such as those with aplastic anemia or myelodysplasia, may tolerate platelet counts as low as 5,000/μL in the absence of complicating factors including fever, infection, and additional defects in hemostasis. 1
Stable chronically thrombocytopenic patients, such as those with aplastic anemia or myelodysplasia, may tolerate platelet counts as low as 5,000/μL in the absence of complicating factors including fever, infection, and additional defects in hemostasis. 1
More aggressive support is indicated for patients who are unstable—febrile, infected, receiving multiple medications—especially if the platelet counts are decreasing.1,12
Platelet transfusions should be monitored by a 1 to 24 hour posttransfusion platelet or complete blood count (CBC) to assess response and guide subsequent transfusion therapy. A corrected count increment (CCI) may be used to determine the increase in platelet count in an individual post platelet transfusion:
![]()
*Posttransfusion platelet count, expressed per microliter, is best obtained 15 minutes to 1 hour posttransfusion
**Body surface area = the square root of [(height in cm × weight in kg)/3,600], expressed in meters squared
***Expressed as multiples of 1 × 1011
An absolute posttransfusion increment of 10,000/μL or greater (approximately 2,000/μL per unit of random donor platelets) in an average-sized adult corresponds to a CCI of 5,000.
Platelet Refractoriness
Patients who respond poorly to repeated platelet infusions are termed refractory. Posttransfusion platelet counts (performed at 1 and 24 hours) are useful tests to determine refractoriness. The CCI should also be calculated and failure to achieve a CCI of 5,000 or greater is cause to suspect platelet refractoriness. Refractoriness may be immune or nonimmune mediated. Immune-mediated refractoriness indicates alloimmunization to HLA or human platelet antigens (HPA).
![]() Nonimmune-mediated causes of platelet refractoriness: fever, infection, splenomegaly, disseminated intravascular coagulation (DIC), massive bleeding, and medications that enhance platelet destruction; more likely to affect the 24-hour posttransfusion count.
Nonimmune-mediated causes of platelet refractoriness: fever, infection, splenomegaly, disseminated intravascular coagulation (DIC), massive bleeding, and medications that enhance platelet destruction; more likely to affect the 24-hour posttransfusion count.
![]() Immune-mediated platelet refractoriness caused by alloimmunization to HLA and human platelet antigens usually associated with multiparity or exposure to nonleukocyte reduced platelet transfusions; more likely to affect the 1 hour posttransfusion count.
Immune-mediated platelet refractoriness caused by alloimmunization to HLA and human platelet antigens usually associated with multiparity or exposure to nonleukocyte reduced platelet transfusions; more likely to affect the 1 hour posttransfusion count.
In practice, the distinction between immune- and nonimmune-mediated platelet refractoriness becomes less clear as alloimmunized patients often have multiple medical issues predisposing to nonimmune refractoriness. When immune-mediated platelet refractoriness is suspected and CCI is less than 5,000 after each of two platelet transfusions the following steps should be taken:
![]() ABO-compatible fresh (less than 72 hours in storage) platelets should be used for two subsequent transfusions
ABO-compatible fresh (less than 72 hours in storage) platelets should be used for two subsequent transfusions
![]() If CCI still does not exceed 5,000, HLA antibody screen to detect alloantibodies, or commercial platelet compatibility tests should be performed
If CCI still does not exceed 5,000, HLA antibody screen to detect alloantibodies, or commercial platelet compatibility tests should be performed
![]() When alloantibodies with broad specificity are found (for HLA A and B loci) platelets from HLA-matched donors are indicated
When alloantibodies with broad specificity are found (for HLA A and B loci) platelets from HLA-matched donors are indicated
![]() Crossmatch compatible platelets may be beneficial when HLA antibody status of the recipient cannot be determined, HLA-matched platelets cannot be obtained, or when the patient is refractory to HLA-matched platelets (up to 40% to 50% of cases)
Crossmatch compatible platelets may be beneficial when HLA antibody status of the recipient cannot be determined, HLA-matched platelets cannot be obtained, or when the patient is refractory to HLA-matched platelets (up to 40% to 50% of cases)
![]() Corticosteroids, washed platelets, or IVIG have not proved useful in the treatment of refractoriness
Corticosteroids, washed platelets, or IVIG have not proved useful in the treatment of refractoriness
Granulocytes
Granulocytes are collected by apheresis for specific patients, from donors who are mobilized prior to collection with corticosteroids and/or granulocyte colony-stimulating factor [G-CSF].
![]() Granulocytes can be stored at room temperature for only up to 24 hours post collection but optimally should be administered within 6 hours of collection.
Granulocytes can be stored at room temperature for only up to 24 hours post collection but optimally should be administered within 6 hours of collection.
![]() Granulocyte collections have a volume of 250 mL and contain plasma, approximately 30 mL RBC, and variable amounts of mononuclear leukocytes and platelets.
Granulocyte collections have a volume of 250 mL and contain plasma, approximately 30 mL RBC, and variable amounts of mononuclear leukocytes and platelets.
![]() Granulocyte concentrates should be ABO, Rh, and RBC crossmatch compatible.
Granulocyte concentrates should be ABO, Rh, and RBC crossmatch compatible.
![]() Products should be irradiated because of the presence of viable lymphocytes in the collection.
Products should be irradiated because of the presence of viable lymphocytes in the collection.
![]() The minimal therapeutic dose is 1 × 1010 granulocytes/unit however, increments are unlikely to be seen unless greater than 3 to 4 × 1010 granulocytes/unit are infused. 1
The minimal therapeutic dose is 1 × 1010 granulocytes/unit however, increments are unlikely to be seen unless greater than 3 to 4 × 1010 granulocytes/unit are infused. 1
Indications: Patients with absolute neutrophil counts of less than 0.5 × 109/L and documented bacterial or fungal infection refractory to antimicrobials. Recipients must have a reasonable expectation of achieving hematopoietic recovery or engraftment (endogenous granulocyte production). Infants with bacterial sepsis, whose granulocyte counts are less than 3 × 109/L with postmitotic neutrophils comprising less than 10% of their nucleated marrow cells, may benefit from granulocyte transfusions.1
A 1 to 6 hour posttransfusion CBC with differential for determination of ANC may help assess efficacy. A 6-hour posttransfusion increment may be higher than a 1-hour posttransfusion ANC because granulocytes travel to the lungs before equilibrating in peripheral blood. If the patient’s ANC fails to reach expected levels or if a reaction occurs, an HLA antibody screen and tests for antibodies to human neutrophil antigens (HNA) are indicated to look for an immunologic cause.
Not indicated: Patients whose bone marrow function is not likely to recover. Contraindicated in patients with prior severe pulmonary reactions to HLA or HNA antibodies or HLA or HNA alloimmunization.
![]() Alloimmunized patients may develop chills, fever, rigors, shortness of breath, wheezing, pulmonary infiltrates, cyanosis, and hypotension6,13; rigors and fever may respond to intravenous meperidine.
Alloimmunized patients may develop chills, fever, rigors, shortness of breath, wheezing, pulmonary infiltrates, cyanosis, and hypotension6,13; rigors and fever may respond to intravenous meperidine.
![]() Pulmonary toxicity may be exacerbated when granulocytes and amphotericin B are administered in close temporal proximity.14 At NIH, amphotericin B administration and granulocyte transfusions are separated by at least 4 hours.
Pulmonary toxicity may be exacerbated when granulocytes and amphotericin B are administered in close temporal proximity.14 At NIH, amphotericin B administration and granulocyte transfusions are separated by at least 4 hours.
![]() Granulocyte transfusion therapy should be evaluated after an initial course of four infusions and then periodically.
Granulocyte transfusion therapy should be evaluated after an initial course of four infusions and then periodically.
![]() Granulocyte concentrates may contain leukocyte-associated pathogens such as cytomegalovirus (CMV), which may be a particular concern for immunosuppressed stem cell transplant recipients, solid organ transplant recipients, neonates undergoing extracorporeal membrane oxygenation, and low birth weight and premature infants.
Granulocyte concentrates may contain leukocyte-associated pathogens such as cytomegalovirus (CMV), which may be a particular concern for immunosuppressed stem cell transplant recipients, solid organ transplant recipients, neonates undergoing extracorporeal membrane oxygenation, and low birth weight and premature infants.
![]() While granulocyte transfusions decrease the length of bacterial infection, proof that granulocyte transfusions decrease mortality in any situation has been elusive. 15,16
While granulocyte transfusions decrease the length of bacterial infection, proof that granulocyte transfusions decrease mortality in any situation has been elusive. 15,16
Fresh-Frozen Plasma
Plasma separated from whole blood or collected by apheresis and frozen within 8 hours is labeled FFP. FFP contains plasma proteins at the time of thaw in about the same concentrations as at the time of collection. The volume of a unit of plasma is approximately 200 mL.
![]() By convention, 1 mL of FFP is expected to provide 1 unit of activity of all factors (except labile factors V and VIII). In practice, individual units may vary in content.
By convention, 1 mL of FFP is expected to provide 1 unit of activity of all factors (except labile factors V and VIII). In practice, individual units may vary in content.
![]() The dose used is 10 to 20 mL/kg in adults (equivalent to approximately 4 to 6 units of FFP) to increase coagulation factor levels by 20%.
The dose used is 10 to 20 mL/kg in adults (equivalent to approximately 4 to 6 units of FFP) to increase coagulation factor levels by 20%.
Indications: Correction of multiple clotting factor deficiencies in patients who are bleeding or prior to an invasive procedure, replacement of factors in consumptive coagulopathy, coagulation factor deficiencies caused by liver disease, dilutional coagulopathy after massive transfusion, replacement fluid for plasma exchange in the treatment of TTP, rapid reversal of warfarin (Coumadin) effect, replacement of single congenital factor deficiencies when no virus-safe fractionated product is available (mostly applies to factor V deficiency).
![]() A PT greater than 1.5 normal or APTT ratio greater than 2.0 in the presence of microvascular bleeding is a guide to consider treatment. 9
A PT greater than 1.5 normal or APTT ratio greater than 2.0 in the presence of microvascular bleeding is a guide to consider treatment. 9
![]() Note, in the setting of life-threatening bleeding associated with Coumadin therapy where rapid reversal is necessary or in overanticoagulated patients with volume overload a more appropriate therapy is Prothrombin Complex Concentrate (PCC). See section Blood Derivatives.
Note, in the setting of life-threatening bleeding associated with Coumadin therapy where rapid reversal is necessary or in overanticoagulated patients with volume overload a more appropriate therapy is Prothrombin Complex Concentrate (PCC). See section Blood Derivatives.
Not indicated: Volume expansion, protein replacement in nutritional deficiencies.
Cryoprecipitate
Cryoprecipitate (cryo) is the cold-insoluble portion of plasma which contains factor VIII, fibrinogen, von Willebrand factor, factor XIII, and fibronectin. Ordinarily stored frozen, cryo can be kept at room temperature for up to 6 hours; on pooling it must be transfused within 4 hours.
![]() Compatibility testing is unnecessary.
Compatibility testing is unnecessary.
![]() A unit of cryo is usually less than 15 mL of plasma and contains more than 80 international units (IU) of factor VIII and more than 150 mg of fibrinogen.
A unit of cryo is usually less than 15 mL of plasma and contains more than 80 international units (IU) of factor VIII and more than 150 mg of fibrinogen.
![]() One unit of cryo can increase fibrinogen in an average adult by 5 to 10 mg/dL.
One unit of cryo can increase fibrinogen in an average adult by 5 to 10 mg/dL.
![]() A therapeutic dose for an adult is 80 to 150 mL of cryo (8 to 10 units pooled).
A therapeutic dose for an adult is 80 to 150 mL of cryo (8 to 10 units pooled).
![]() Indications: Treatment of congenital fibrinogen deficiency, dysfibrinogenemia, factor XIII deficiency, DIC (if fibrinogen <1.0 g/L).
Indications: Treatment of congenital fibrinogen deficiency, dysfibrinogenemia, factor XIII deficiency, DIC (if fibrinogen <1.0 g/L).
![]() Note a pathogen-inactivated fibrinogen concentrate has recently become available in the United States for treatment of congenital fibrinogen deficiency.
Note a pathogen-inactivated fibrinogen concentrate has recently become available in the United States for treatment of congenital fibrinogen deficiency.
Cryo has also been used to correct the platelet defect of uremic bleeding, although with variable success.
The dosage of cryo depends on the underlying deficiency and on the plasma volume of the patient. To determine the number of bags of cryo to replace fibrinogen
![]()
*The plasma volume for an average adult = (1 {{{727}}} percent hematocrit/100) × patient weight in kg × 70 mL/kg. For infants and children under 40 kg, the plasma volume = (1{{{727}}}percent hematocrit/100) × patient weight in kg × 80 to 85 mL/kg.
Not indicated: Factor VIII deficiency and von Willebrand disease for which more specific and safer products now exist.
Hematopoietic Stem and Progenitor Cells
Optimal outcomes in HSCT depend on the successful procurement of cells from patients (autografts) or donors (allografts). Several recent developments in this field have improved clinical outcomes making this approach a safe and effective therapy for a variety of malignant and nonmalignant disorders.
Stem cell sources now include related and unrelated bone marrow, peripheral blood and umbilical cord blood. The vast majority of HSCT now uses mobilized peripheral blood although guidelines for management of aplastic anemia still recommend using a bone marrow source if possible.17
Historically, mobilization of stem cells into peripheral blood for autografting was done using myelosuppresive chemotherapy such as cyclophosphamide as increased numbers of cells entered the circulation during the recovery phase of the bone marrow. In the late 1980s G-CSF and GM-CSF became available and were used either alone or in combination with chemotherapy as mobilizing agents. G-CSF is now the standard for this indication.
A significant proportion of patients do not mobilize with G-CSF with or without chemotherapeutic agents. Factors that have been shown to predict for poor mobilization include increasing numbers of cycles of prior chemotherapy, prior radiation therapy, and the presence of marrow metastasis.18–20 Fludarabine is particularly toxic to stem cells and this should be avoided in patients for whom a stem cell collection for autograft is planned. It has also been shown that there is a correlation between the number of circulating CD34+ cells and the probability of obtaining an adequate collection for transplant.21
Until recently poorly mobilizing patients had few options. However, an agent originally developed to treat HIV, plerixafor, has recently been shown to have significant activity in these patients. In contrast to G-CSF which needs multiple injections over several days a single dose of plerixafor mobilizes stem cells into the periphery beginning at 1 hour and peaking at 10 hours.22 In addition the quality of the stem cell graft mobilized by plerixafor may be superior. Compared to G-CSF mobilized cells, the plerixafor-mobilized cells were more primitive and therefore more quiescent and produced superior engraftment in both NOD/SCID mice and human recipients.23 The combination of G-CSF and plerixafor has also been studied and produces greater increases in CD34+ cells than with either agent alone.24
Standard collections of allogeneic peripheral blood stem cell (PBSC) involve 3 to 4 hours per apheresis procedure, during which approximately 10 L of blood are processed. At least two collections are usual, but large volumes of 25 to 30 L are more efficient and used increasingly to allow complete collections with a single procedure.25 Donor demographic and laboratory predictors such as platelet count and CD34+ mononuclear cell count can be used to customize the length of the collection procedure.26
As G-CSF is still the standard agent used, complications seen with stem cell mobilization are often related to this cytokine. Bone pain, headache, fatigue, insomnia, and gastrointestinal disturbances are usually mild and respond to administration of acetaminophen or NSAIDs. Spleen size increases in almost all individuals on G-CSF and this has been associated with splenic rupture. Donors should be advised to refrain from contact sports for a few weeks after the last mobilization.27 Vascular complications and citrate toxicity are not unlike those experienced in other long apheresis procedures (see below).
PBSC grafts are infused as fresh collections or stored frozen with the cryoprotectant dimethyl sulfoxide (DMSO) in liquid nitrogen. Thawed cells infused with DMSO may cause nausea, vomiting, fever, dyspnea, hypotension, and anaphylaxis. Reactions are dose dependent and may be lessened by prophylactic antihistamines. PBSCs carry the risk of transfusion-transmitted infectious agents and are tested in the same manner as are other blood components. However, given their highly specialized use and their life-saving potential, exceptions are made to donor selection criteria normally used for allogeneic blood collections, with the concurrence of the treating physician and the recipient.
Adequate cell dose for engraftment depends on whether the procedure is an autograft, a related, or an unrelated allograft. Cell dose, cell source, and patient characteristics are all important variables. The dose of stem cells for unrelated donors (National Marrow Donor Program) is 2 to 4 × 108 nucleated cells per kilogram of recipient weight, with 2 to 4 × 106 CD34+
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


