The most reliable results for the detection and identification of red cell antibodies are obtained by using a combination of tests. The particular combination of tests adopted in specialist blood group reference laboratories depends on such factors as the nature of the problems to be solved, including the urgency with which a result is needed, the technical expertise of the staff available and financial restraints.
In identifying red cell antigens, the test used depends on the characteristics of the antibodies available. The simplest technique is agglutination in saline, and this test can almost always be used when the relevant antibody is IgM. When the antibody is IgG it will usually be necessary to use some slightly more complicated procedure (commonly, addition of antiglobulin reagent or pre-treatment of red cells with proteolytic enzymes). A solid-phase system can also be used. In this case antibody is bound to a surface and the adherence of red cells (carrying the antigen) measured. The gel test can also be used for red cell antigen typing and involves incubating the antibody with red cells on top of a dextran gel and then distinguishing agglutinated red cells from unagglutinated cells by centrifugation.
Gel test and solid-phase methods are increasingly used in routine blood banking in preference to traditional agglutination methods because they are easier to perform and less dependent on the technical competence of the individuals performing the test. Monoclonal antibodies (see Chapter 3) have largely replaced polyclonal antibodies as reagents for routine blood grouping procedures based on agglutination.
The reaction between red cell antigens and antibodies may be detected in various other ways: for example, the antibody may be labelled with a radioisotope such as 125I and the amount of 125I bound to the cells measured. The antibody may be coupled to an enzyme and the binding of antibody to red cells measured by the amount of colour released from a suitable substrate, as in the enzyme-linked immunosorbent assay (ELISA). The antibody may be coupled to a fluorescent dye and the binding of antibody to red cells measured by its bound fluorescence using flow cytometry. When an antibody to a blood group antigen is labelled in one of these ways the test can be used to quantify the number of antigen sites per red cell in the sample examined or to compare the amount of antigen expression on red cells from different samples. However, it is more usual to label an anti-IgG preparation with radio-iodine, enzyme or fluorescein and to use this reagent for quantification of antigen sites or blood group antibody molecules bound, as in this case only one labelled antibody preparation is required to quantify the binding of many blood group antibodies.
The object of most blood grouping and antibody detection tests in clinical work is to provide completely compatible red cells for transfusion; the only question to be answered is: is a particular antigen or antibody absent or present? Quantitative tests are needed in assessing the significance of antibodies found in women in connection with pregnancy.
Blood typing can also be carried out using methods based on detection of the DNA sequence encoding blood group antigens rather than agglutination. These methods are particularly useful for determination of the blood group of a fetus at risk from haemolytic disease or neonatal anemia (see Chapter 12). Molecular methods can also be used to find antigen-matched donor red cells for patients with complex mixtures of antibodies when the typing reagents to the less common antigens, which are needed to screen for compatible blood by agglutination methods, are in short supply (see ‘Molecular methods of red cell grouping’).
Quantitative Differences between Red Cells from Different Donors
Homozygotes and Heterozygotes
With respect to some red cell antigens, significant differences in reactivity are found between the cells of homozygotes and heterozygotes. Some examples are as follows: C, c, M, S K and Jka. This is an important consideration when selecting red cells for use in screening sera for the presence of blood group antibodies, as clinically significant antibodies may be missed when cells heterozygous for important antigens like K and Jka are used (Phillips and Voak 1996). In 1997, 95.5% of participants in the UK National External Quality Assessment Scheme were selecting screening cells that had homozygous expression of antigens (Knowles et al. 2002). DNA-based methods of blood group typing provide a useful additional tool for the selection of cells homozygous to certain alleles like Doa/Dob where suitable antibody reagents are scarce or where homozygosity cannot reliably determined using serological methods (RhD and Fy zygosity, for example, Hult et al. 2005).
Differences between Donors of the Same Phenotype
The reactivity of red cells from different individuals of the same phenotype is particularly variable with regard to the following antigens: P1, I, i, Lea, Leb, Sda, Vel and Ch/Rg. This property is a useful aid to the identification of antibodies of these specificities.
Differences between Newborn Infants and Adults
For details, see Chapter 3.
Effect of Storage on Red Cell Antigens
Storage Above 0°C
When red cells were stored at 4°C either as whole blood mixed with CPDA-1 or -2, or with saline–adenine–glucose (SAG) additive for up to 35 or 42 days, respectively, very little loss of activity of red cell antigens other than M and P1 was observed (Snyder et al. 1983; Myhre et al. 1984). Gradual loss of expression of the complement regulatory proteins DAF (Cromer blood group protein) and CD59 from stored packed red cells was observed over a period of 6 weeks by Long and co-workers (1993). Red cell microvesicles derived from outdated units of red cells have A, B, D, M, N, Ss and Fya activity as determined by adsorption studies (Oreskovic et al. 1992).
Red cells stored as clotted blood lose their antigenic activity more rapidly than when stored with citrate anticoagulant (Rosenfield et al. 1971). When blood is collected into plastic bags, if the donor line is not emptied immediately after collection and then refilled with blood mixed with anticoagulant, the clotted blood in the tubing is an unreliable source of red cells for compatibility tests (Jørgensen 1964). Furthermore, red cells stored as clotted blood may give false-positive reactions in the antiglobulin test owing to uptake of complement components during storage at 4°C.
Reagent red cells may be stored as whole blood with citrate–phosphate–dextrose (CPD) or acid–citrate–dextrose (ACD) but more usually they are stored as washed cells in a preservative solution. A modified Alsever’s solution, with added inosine (with or without adenine) and with antibiotics is commonly used, permitting satisfactory storage for at least 35 days at 4°C.
Before being suspended in preservative solution, red cells should be freed, as far as possible, from leucocytes. Leucocytes contain proteolytic enzymes that, in the absence of plasma inhibitors, may cause lysis of red cells (Högman et al. 1978). In red cell suspensions stored in LISS in the presence of neomycin, proteolytic enzymes from leucocytes may damage red cell antigens. The damage occurs only when leucocytes are present in combination with LISS and neomycin (Malyska et al. 1983). The exact mechanism responsible for the damage is not known, but aminoglycoside antibiotics may promote the release of proteolytic enzymes from leucocytes. Damage to antigens was found to be much more serious with aminoglycoside antibiotics, such as neomycin, than with others (Allan et al. 1990). Red cells stored in LISS to which amphotericin B has been added are lysed rapidly, probably due to an increased susceptibility of red cells in LISS for sodium deoxycholate present in the amphotericin solution. However, frozen-thawed red cells can be stored safely for 21 days at 4°C in a LISS solution that does not contain either aminoglycoside antibiotics or amphotericin B (Allan et al. 1990). The following solution is recommended: glycine, 13.7 g; glucose, 8.5 g; NaCl, 1.9 g; Na2HPO4, 0.21 g; NaH2PO4.2H2O, 0.23 g; adenine, 0.2 g; inosine, 0.4 g; chloramphenicol, 0.34 g and water to 1 l.
Frozen Storage
When red cells were stored at −20°C in a citrate–phosphate–glycerol solution (see Appendix 12) for 1 year the only antigen found to react more weakly was P1 (Crawford et al. 1954). After thawing the frozen cells, glycerol must be removed before the cells can be tested; removal is most conveniently carried out by washing; see Appendix 12.
Unfortunately, when red cells are stored at −20°C there may be extensive haemolysis, possibly dependent on the time for which the red cells were stored before freezing.
For storage for indefinite periods red cells must be kept at −80°C or lower. One very convenient method is known as ‘glycigel’. Tubes containing a glycerol solution mixed with gelatin are kept available in the refrigerator. When red cells for storage are added to one of these tubes and the tube is warmed, the glycerol is released slowly as the gelatin melts and cell damage is minimized. The red cells are stored at −80°C or lower and, when wanted, are thawed and washed free from glycerol (Huggins et al. 1982; see also Appendix 11).
A very simple method for preparing red cells for frozen storage is to add dextrose or sucrose to them and then add the mixture dropwise from a syringe into liquid nitrogen (Meryman 1956). Another method that has proved very satisfactory is the one described by Bronson and McGinniss (1962). Blood mixed with an equal volume of Alsever’s solution is mixed with one-tenth of its volume of 50% glucose; a metal screen is dipped into the mixture, which is then frozen by immersion in liquid nitrogen. After storage in nitrogen vapour, the blood is thawed by placing the screen in a tube containing 5% glucose in saline at 40°C. The cells are then washed. In washing red cells that have been frozen in this way, better results are obtained if the thawed cells are first washed in a hypertonic solution (Burnie 1965). An alternative excellent method for reagent red cells utilizes polyvinyl pyrrolidone (PVP). PVP (molecular weight 44 000) is prepared as a 30% solution (w/v) in water, autoclaved, mixed well and stored in aliquots. Three volumes of PVP solution are added to 10 volumes of anticoagulated blood (or 45% suspension of red cells in PBS, pH7.4). The mixture is then added, dropwise, to liquid nitrogen. The frozen beads obtained are stored in a pre-chilled container in nitrogen vapour. To recover the cells, drop the beads into warm phosphate-buffered saline (PBS) pH 7.4 (approximately 2 ml of PBS per bead), centrifuge and wash three times with PBS (G Inglis, personal communication).
Freeze–thaw damage to red cells can be reduced using ice-binding proteins found in Antarctic sea ice diatoms (Kang and Raymond 2004).
Lyophilized Red Cells
The disaccharide trehalose functions to reduce dehydration in plants and animals living in desert habitats and can be used to reduce osmotic damage in stored red cells. Trehalose has been used for stabilization of dried red cell-coated microplates and may ultimately facilitate storage of red cells by lyophilization (Thorpe et al. 2002; Satpathy et al. 2004).
When whole units of blood are needed to provide cell panels, any method for the frozen storage of blood for transfusion is suitable (see Chapter 9).
Factors Affecting Red Cell–Antibody Interactions
Serum Vs. Plasma
For antibody detection, plasma has the theoretical disadvantage, compared with serum, of containing anticoagulants that inhibit complement activation and therefore possibly interfere with the detection of some complement-activating antibodies. On the other hand, plasma is more convenient and is widely used in automated systems of grouping and antibody screening. A comparison of plasma and serum for antibody detection using the gel system has been reported (Scott et al. 1996).
Storage of Sera
If there is no microbial contamination, at 4°C blood grouping sera retain their potency for 1–2 years; when stored at −20°C or less, they retain their potency for very many years.
The use of sodium azide is potentially hazardous. When solutions containing azide are discharged to waste via metal pipes, heavy metal azides, notably of copper or lead, are formed, which are readily detonated, explosive compounds. Copper azide is particularly sensitive to mechanical shock. Despite these potential hazards, sodium azide is still added to most commercial blood grouping reagents. Antibiotics such as those used in red cell suspensions may be used as a substitute for azide but corresponding antibodies are sometimes present in human sera (see Chapter 7) and then produce anomalous results.
Colouring Agents
Dyes are frequently added to anti-A and anti-B reagents; usually blue is added to anti-A and yellow to anti-B; Patent blue V (E131) and Ariavit tartrazine (E102) in concentrations of 0.08 g/l are suitable. Although colour is convenient in identifying reagents, it must not be relied on, i.e. the reagent should be identified by reading the label, and the use of controls remains essential. A green dye is added to some polyspecific antiglobulin reagents.
Deterioration of Complement on Storage
When serum with an optimal concentration of complement is required it should be separated from red cells as soon as possible (Fischer et al. 1958). On storage of serum at 4°C there is just-detectable deterioration of complement after 24 h and readily detectable deterioration at 1 week (Polley and Mollison 1961).
Garratty (1970) found that over 60% of normal complement activity was required to avoid the risk of missing weak complement-binding antibodies. In a study of normal sera this level of activity was retained for less than 1 day at 37°C, for 1 day at room temperature, for 2 weeks at 4°C, for 2 months at −20°C and for at least 3 months at −55°C or below (see also Polley and Mollison 1961). Complement deteriorates far less rapidly in the plasma of ACD stored blood than in serum, possibly due to the diminished levels of Ca2+ and Mg2+; after three weeks at 4°C the average loss of haemolytic activity is only 9%. If ACD plasma is used as a source of complement, it must be recalcified (Reich et al. 1970).
Potentiators
The effect of colloids in potentiating agglutination is discussed in Chapter 3.
Polyethylene glycol (PEG) is used as a potentiator in the antiglobulin test (see below), but other substances, such as dextran and PVP, are no longer used in manual tests.
Proteolytic Enzymes
As discussed in Chapter 3, treatment of red cells with certain enzymes renders them agglutinable by otherwise non-agglutinating antibodies.
Several proteolytic enzymes are used in blood grouping tests: trypsin and chymotrypsin, which have a very restricted specificity, and bromelin, ficin and papain, which have a broad specificity for peptide bonds. Trypsin cleaves peptides that are C-terminal to lysine and arginine, whereas chymotrypsin catalyses cleavage primarily after the C-terminal of phenylalanine and tryptophan. Bromelin, ficin and papain are all thiol proteases with a cysteine residue at the active site, which must be in its reduced form for activation of the enzyme. Thiol proteases are readily inactivated under normal storage conditions as a result of oxidation of the thiol group of the cysteine or by binding of traces of heavy metal ions to the thiol group. Optimal activation of thiol proteases occurs on simultaneous addition of a thiol reducing agent, such as cysteine, and a heavy metal chelating agent such as EDTA (Kimmel and Smith 1954). Reducing agents have a greater effect on activity than do chelating agents (Scott et al. 1994).
Traditionally, in blood group serology, papain has been used in its activated form (for example Low’s formulation containing cysteine and sometimes EDTA), whereas bromelin and ficin have been used inactivated. The activity of bromelin and ficin can be greatly increased by the addition of sulphydryl reducing agents (Scott et al. 1994).
Enzyme preparations should be standardized either by using azoalbumin as a substrate (Lambert et al. 1978; Scott and Whitton 1988), or by comparison with a standard enzyme preparation, using a standard blood grouping reagent. A lyophilized preparation of papain at 0.6 azoalbumin units and a lyophilized reference anti-D have been prepared by the International Society of Blood Transfusion (ISBT)/International Committee on Standardization in Haematology (ICSH) Working Party on Enzymes (Scott et al. 1994). In practice, enzyme preparations are frequently assessed for potency by subjective serological tests.
Effect of Enzymes on Particular Red Cell Antigens
In the detection of Rh antibodies, tests with enzyme-treated red cells are extremely sensitive. Giles (1960) found that additional Rh antibodies were often revealed by tests with ficin-treated red cells: for example, ‘pure’ anti-D might be found to contain anti-C or anti-E, ‘pure’ anti-C to contain anti-e and ‘pure’ anti-E to contain anti-c.
The reactions of anti-P1 and of Lewis and Kidd antibodies are stronger with enzyme-treated than with untreated red cells and Lewis and Kidd antibodies may lyse enzyme-treated red cells when they will not lyse untreated cells. Other antibodies that react more strongly with enzyme-treated than with untreated cells include anti-I and -i and the antibodies of the Colton and Dombrock systems.
Antigens inactivated or weakened by treatment with certain proteolytic enzymes include Fya, Fyb, M, N, S, s, Yta, Xga, Ge, Ch, Rg, JMH, Inb, Knops system antigens, Cromer system antigens, Tn and Pr (Table 8.1). Crude preparations of bromelin, ficin and papain are mixtures of many proteolytic enzymes and these three enzyme preparations weaken most of the above antigens. On the other hand, crystalline trypsin and chymotrypsin have more selective effects: S, s and Fya are destroyed by chymotrypsin but not by trypsin, and K is only destroyed when both enzymes are used together; some antigens, for example M, N and Lua, are destroyed by either enzyme (Judson and Anstee 1977).
Table 8.1 Effect of various proteases, sialidase and AET on high-frequency red cell antigens.
Source: Daniels G 1992. Reproduced with permission of the American Red Cross.
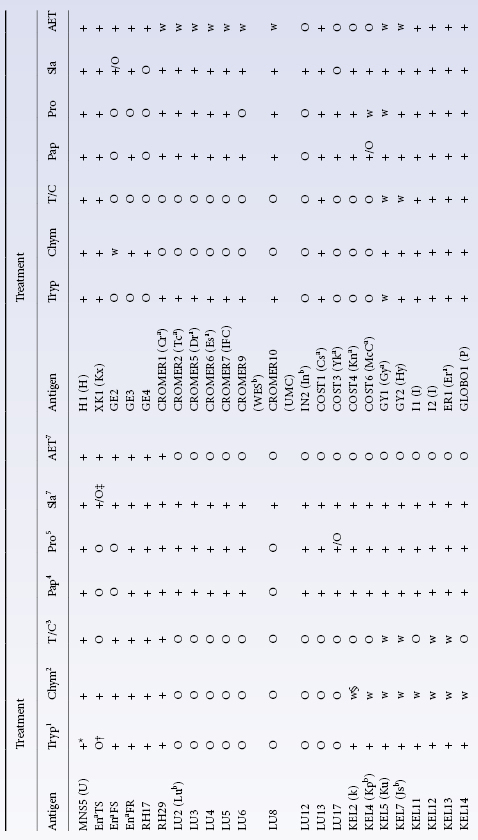
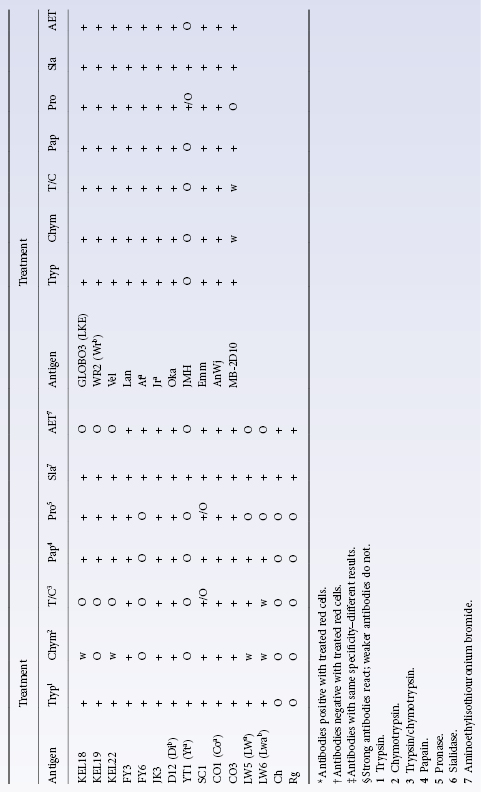
Lua and Lub were found to be unaffected by treatment with ficin or papain by G Garratty (personal communication), although variable effects were found by Poole and Giles (1982).
The low-frequency antigens of the MNSs system can be classified according to their sensitivity to trypsin treatment. The trypsin-sensitive determinants are located on glycophorin A, carrying MN determinants, whereas the trypsin-insensitive determinants are probably located on glycophorin B, carrying Ss determinants, although they may be located on glycophorin A close to the red cell membrane (Giles 1982).
The reactions of anti-Yta are not diminished by trypsin treatment of red cells but, in one series, six of six examples failed to react with cells treated with bromelin, chymotrypsin, ficin or papain (Rouger et al. 1982) and in another series, 8 out of 14 examples of anti-Yta failed to react with cells treated with ficin or papain (Vengelen-Tyler and Morel 1979).
The determinants Pr, Tn and T are weakened by treatment with proteolytic enzymes but are not inactivated. Some activity persists because the determinants occur not only on glycophorins A, C and D, which are split off the red cell membrane by most proteolytic enzymes, but are also carried on glycophorin B, which is not affected by treatment with most proteolytic enzymes (the exception being chymotrypsin).
In some cases, the antigens are damaged only by relatively high concentrations of enzyme. For example, in one investigation destruction of s was not observed until chymotrypsin levels of 132 units/ml were used and even at this level the receptor detected by one example of anti-S was unaffected (Judson and Anstee 1977). Similarly, although T is not destroyed by concentrations of papain normally used in blood grouping tests (Bird et al. 1971) it is destroyed by 3–4% ficin or papain (Issitt et al. 1972).
Stability of Enzymes on Storage
Cysteine-activated papain stored at 4°C as a liquid at pH 5.4 loses about 50% of enzyme activity within 2 weeks; most of this loss is due to instability of the activator (ML Scott and PK Phillips, personal communication). Preparations that have not been cysteine activated are relatively stable during storage at 4°C (Stapleton and Moore 1959). Because of the instability of activated enzyme preparations, which may be the cause of false-positive or false-negative results (Holburn and Prior 1987), many users prepare their own enzyme solutions and store them at −20°C, at which temperature they may be kept for 6 months.
Liquid enzyme preparations can be stabilized in a form suitable for distribution by reversible inhibition of the enzyme by heavy metal ions; activated enzyme is recovered by the addition of a separately provided activator (ML Scott and PK Phillips, personal communication). As an activator, cysteine is unsuitable because of its instability; dithiothreitol (DTT) and glutathione are both stable on storage at 4°C but, as DTT added to activated papain destroys Kell antigens (Branch and Petz 1982), glutathione is preferred.
Low Ionic Strength
As described in Chapter 3, the rate of association of antibody with antigen is very greatly increased by lowering ionic strength. The advantages of using a low-ionic-strength medium are seen chiefly in tests with IgG antibodies (see section on antiglobulin tests, below). Nevertheless, it is a fairly common practice to use a low-ionic-strength medium instead of saline for the suspension of red cells. Most workers have found that with anti-A and anti-B results are similar with low-ionic-strength medium and with saline (Elliot et al. 1964; Löw and Messeter 1974; Moore and Mollison 1976). The reactivity of some cold alloagglutinins is enhanced in a low-ionic-strength medium so that their thermal range becomes wider, for example anti-A1 and anti-P1 (seventh edition, p. 519); others, for example two examples of anti-Prl are detectable only in a low-ionic-strength medium (O’Neill et al. 1986). The enhanced activity of cold alloagglutinins may complicate blood grouping and pre-transfusion testing if low-ionic-strength techniques are used in direct agglutination.
Confusion is caused by using the term ‘low ionic strength’ to describe a variety of solutions supplied with reagent kits. These may be used under the impression that their composition is identical with some of the earlier described solutions (e.g. Moore and Mollison 1976), although in fact, they may have a higher tonicity.
Polycations
Protamine and polybrene cause red cells to aggregate. The resulting very close contact allows crosslinking of red cells by IgG antibodies. Non-specific aggregation can be dispersed by the addition of a suitable agent and then only specific agglutinates remain. Manual tests in which a low-ionic-strength medium is used together with protamine have proved very sensitive in detecting alloantibodies. If no agglutination occurs the cells can be washed and antiglobulin serum added as a further step (Rosenfield et al. 1979; Lalezari and Jiang 1980). Details of a manual polybrene test (MPT) are given later.
Monoclonal Vs. Polyclonal Antibodies
Monoclonal antibodies have several advantages over polyclonal antibodies: (1) they provide an unlimited supply of identical material; (2) they are free from unwanted contaminating antibodies, for example HLA antibodies or antibodies to low-frequency antigens, so that testing and quality control are far simpler; (3) they are free from naturally occurring antibodies such as anti-T so that they do not give false-positive results, for example with T-transformed red cells; (4) they are free from viruses of hepatitis and of HIV; and (5) their use avoids the need to immunize and plasmapherese human donors.
Murine monoclonal antibodies have at least two disadvantages: first, as their specificities may apparently vary at different concentrations, tests employing them must be very carefully standardized (MacDonald and Gerns 1986); second, they may give unexpected reactions, for example some potent monoclonal anti-A react weakly with some B cells and some potent anti-B react with A1 cells (see Chapter 4). Other possible disadvantages of monoclonal antibodies are mentioned in Chapter 3.
Antibodies of only certain specificities can be produced in mice. Some excellent human monoclonals have been produced (see Chapters 3, 4, 5 and 6). Monoclonal antibodies can be converted from IgG to IgM using recombinant DNA technology in order to make them more suitable for use as blood typing reagents. Lee and co-workers (2004) describe the production and use of humanized chimeric IgM monoclonal anti-Fya and anti-Jsb. Dimeric Fab of anti-N and anti-M specificities have been produced using recombinant techniques and expression in Escherichia coli. These dimeric Fabs had similar agglutinating properties to the corresponding bivalent IgG antibodies (Czerwinski et al. 2002). (Polyclonal anti-M and anti-N are usually of the IgG class and are direct agglutinins because the M, N antigens are located at the amino terminus of glycophorin A, some distance from the lipid bilayer, enabling IgG to bridge two red cells in suspension; see Chapter 3.)
Reduced and Alkylated IgG Antibodies
As described in Chapter 3, after mild chemical reduction and alkylation IgG3 incomplete antibodies will agglutinate red cells suspended in saline. Agglutination is greatly enhanced by adding polymerized BSA to a final concentration of 6–8%. Reagents containing reduced anti-D have been superseded by monoclonal IgM anti-D.
Agglutination Tests
In the agglutination reaction two separate processes may be recognized: first, the uptake of antibody onto the red cells and second, the adherence to one another of the antibody-coated cells. In agglutination tests, the red cells may be allowed to come into contact with one another by sedimentation or the process may be accelerated by the use of centrifugation.
Tests with Untreated Red Cells in Saline
Need to Wash Red Cells
For traditional serological tests, red cells should be washed free of their own plasma. If this is not done, clots will form when the red cells (contaminated with fibrinogen) are mixed with serum, which contains residual thrombin. Other reasons for washing red cells are as follows: (1) plasma tends to cause rouleaux formation, which interferes with the interpretation of agglutination tests; (2) plasma contains anticoagulants that are anti-complementary and may thus interfere with the detection of complement-binding antibodies; (3) various substances added to red cell suspensions, for example lactose or neomycin, are occasionally responsible for agglutination when corresponding antibodies are present in the patient’s plasma (see Chapter 7); most of the antibodies concerned do not react with red cells washed in saline; (4) ABO, Lewis, Chido/Rodgers and Ii antigens are present in the plasma and may inhibit the corresponding antibody in the test serum; and (5) plasma may contain so-called albumin autoagglutinins which used to cause false-positive reactions when whole blood was added to a serum albumin mixture.
Optimal Period of Incubation
The optimal period is determined first by the rate of combination of antibody with red cells and, second, by the time taken for antibody-coated cells to come into contact with one another. Factors affecting the uptake of antibody, namely the ratio of serum to cells, the period of incubation, temperature and the tonicity of the medium, are discussed in Chapter 3. Contact between antibody-coated cells occurs relatively slowly if the red cells are simply left to sediment in a column, for example in a test tube. Alternatively, the cell suspension can be centrifuged.
Tests in Tubes
Tubes in racks can easily be placed in a waterbath (at 37°C), incubator or heat block. The period of incubation is usually 45 min. The red cells can be allowed to sediment or the tubes can be centrifuged. The tube is then tapped and rolled to resuspend the cells. If no agglutinates are seen with the naked eye the tube can be examined with a hand lens, over a magnifying mirror or under a low-power microscope.
Microplates (Microtitre Plates)
Microplates are clear plastic plates containing, as a rule, 96 wells (8 × 12). With V-shaped wells, very weak cell suspensions (0.03%) can be used, but with U-shaped wells, 1–2% suspensions are optimal. One small volume (e.g. 20 µl) of serum is added to an equal volume of red cell suspension. The plates are centrifuged. As a rule, U wells are agitated and read in a conventional way. V wells are tilted at 75°C to the horizontal for 10 min. Unagglutinated cells stream down the side of the well as a smooth thin line; agglutinated cells remain as a button. The use of microplates has several advantages: the method is very much more sensitive than that of other agglutination systems, primarily because very weak cell suspensions can be used; very small amounts of reagents are needed; titrations are easier with multi-channel pipettes and grades of reactions can be compared. A problem with microplates is that some antibody reagents cannot be used undiluted because their high viscosity causes red cells to adhere to the side of the wells. This problem arises particularly when potentiated anti-D reagents and, to a lesser extent, when anti-A and anti-B reagents are used. The problem can be overcome by diluting the reagents with saline. Some monoclonal antibodies adhere to the plastic solid phase and a positive reaction is seen as a monolayer of cells, whereas a negative reaction is seen as a button or stream. A semi-automated system is mentioned in a later section. For a review of the use of microplates, see Blood Transfusion Task Force (1991).
Slide Tests
Slide tests should be used only if equipment for other tests is not available. As water evaporates rapidly from the large surface area, slide tests must be read within 5 min or so; in practice, reagents that produce strong agglutination within 1–2 min are normally used and the tests are employed simply for rapid determination of ABO and RhD groups. As the results are read macroscopically, strong cell suspensions (20% or more) should be used to facilitate the detection of agglutination.
Tests in Capillary Tubes
A method of Rh grouping in capillary tubes was described by Chown (1944) and Chown and Lewis (1946; 1951). Two important advantages of the method are that each test required only 5 µl of serum and that results can be read in 15 min. For a review of the many applications of capillary tube tests, see Crawford et al. (1987).
Methods Using Proteolytic Enzymes
One- Vs. Two-Stage Methods
One-Stage Method
In a method described by Löw (1955), enzyme (cysteine-activated papain), serum and cells are mixed together, thus allowing enzyme treatment of the cells and the reaction of antigen and antibody to occur simultaneously. Evidently, in such a method an opportunity is provided for the cleavage of Ig molecules by the enzyme. The proteolytic effect of the enzyme is diminished by protease inhibitors in serum (Travis and Salvesen 1983) but such inhibitors also reduce the effect of enzyme on the red cells. Although one-stage methods are sufficiently sensitive to give reliable results with potent blood grouping reagents, they are unsuitable for antibody detection or determination of specificity with manual techniques.
Two-Stage Method
In this method, washed red cells are first treated with an enzyme, then washed again and incubated with serum. The method has two substantial advantages over a one-stage method: first, no opportunity is provided for degradation of Ig by enzyme; second, enzyme treatment and red cell–antibody interaction can each be carried out at its optimal pH. For papain and bromelin treatment of red cells, the optimal pH is 5.4–5.8, as determined by serological results in a two-stage technique (Scott et al. 1987). The optimal pH for binding of anti-D to red cells is 6.5–8.0 (Hughes-Jones et al. 1964a), although the optimum for antibodies of other specificities may be different.
A two-stage papain technique in which washing of the cells after papain treatment is replaced by the addition of a specific papain inhibitor, E-64, has been described by Scott and Phillips (1987). This technique permits optimal enzyme treatment of the red cells while avoiding digestion of immunoglobulins following the addition of serum. It thus combines the sensitivity of two-stage tests with the convenience of one-stage tests. The inhibitor E-64 can also be used for bromelin (Ogasawara and Mazda 1989).
In automated blood grouping machines, red cells are suspended in enzyme solution and incubated briefly before being mixed with the sera.
A Comparison of One- and Two-Stage Methods
In a study by the ISBT/ICSH Working Party on Enzymes, the sensitivities of a two-stage method and of various one-stage methods, namely one-stage mixing (Löw 1955), one-stage layering (Dodd and Eeles 1961), a one-stage phase technique (Odell et al. 1983) and the phased inhibitor technique (Scott and Phillips 1987) were compared. Only the two-stage technique was found to be sensitive enough to be used for the detection of antibodies. When papain or bromelin was used at 0.6 azoalbumin units, the two-stage technique was slightly more sensitive than the standard LISS spin-tube IAT (Scott et al. 1994). A disadvantage of the two-stage technique is that false-positive reactions occur when the enzyme activity is too great. Use of the ISBT/ICSH reference preparation should allow the standardization of enzyme preparations and techniques in individual laboratories and thus avoid false-positive results.
The clinical significance of antibodies detectable only with enzyme-treated red cells (‘enzyme-only antibodies’) has been much discussed. As described in Chapter 10, such antibodies are sometimes capable of causing accelerated destruction of small volumes of incompatible red cells, but they seem hardly ever to cause clinically significant haemolysis. In a retrospective study on serum samples from 10 000 recently transfused patients, 35 were found to contain enzyme-only antibodies. In total, 19 out of the 35 patients had been transfused with incompatible red cells and only one had developed a DHTR. However, in 17 of these 19 cases the antibody was anti-E and, in most cases at least, may well have been naturally occurring. In the remaining two cases, the specificities were anti-c and anti-e and in the first of these a DHTR developed (Issitt et al. 1993).
As tests with enzyme-treated cells detect many clinically insignificant antibodies, they should not be used in pre-transfusion testing. On the other hand, enzyme-only antibodies may be found in the early stages of primary immunization, for example in pregnancy and in patients (e.g. those with thalassaemia) receiving regular transfusion.
Role of Ionic Strength
The effects of low ionic strength and of enzyme treatment of cells are not additive (Atchley et al. 1964; Elliot et al. 1964). In external quality assessment surveys the use of low ionic strength was found to have no influence on results obtained with enzyme techniques (Holburn and Prior 1987).
The Manual Polybrene Test
As discussed in a later section, protamine and polybrene are used in certain automated techniques for antibody detection; they are also used in manual tests. In the low-ionic polycation test, protamine is used and in the manual polybrene test (MPT), polybrene. Of these two tests, the most satisfactory appears to be the MPT, as it requires fewer manipulations and only a very brief incubation period at room temperature.
One millilitre of a low-ionic-strength medium (dextrose-EDTA) is added to two drops of a 10% cell suspension and two drops of serum; after 1 min at room temperature, two drops of 0.05% polybrene are added, the tubes are spun, and the supernatant is decanted. Two drops of a citrate–dextrose solution are added to disperse polybrene-induced aggregation and the contents of the tube are mixed by gentle rolling. After examining the tube for agglutinates, the red cells can be washed and the antiglobulin test carried out. As C3 and C4 may bind non-specifically at low ionic strength, the antiglobulin reagent should not contain anticomplement (Lalezari and Jiang 1980).
A modified test in which two drops of serum are mixed with one drop of a 3% suspension of red cells and 0.6 ml of low-ionic-strength medium was used by Ferrer and co-workers (1985). This test was found to be more sensitive than the original test described by Lalezari and Jiang (1980).
For the detection of most antibodies, the MPT was found to be more sensitive than either the standard two-stage enzyme method or the IAT (Fisher 1983). However, anti-K and anti-Fya (particularly the former) are often missed, and may be missed even when the MPT test is followed by a test with antiglobulin serum (Fisher 1983; Ferrer et al. 1985; Malde et al. 1986). Using a commercial kit for the MPT, 31 of 47 anti-Ks were not detected and this MPT test was therefore considered to be unsuitable as a primary technique for the detection of alloantibodies (Letendre et al. 1987).
For large-scale red cell typing the MPT was found to be as good as routine microplate testing but twice as quick. Antiglobulin serum was needed only for Kell grouping (Etges et al. 1982). Satisfactory results were observed in another series, although antiglobulin serum was used for testing with anti-Jka, -S and -s as well as with -K (Anderson and Patel 1984). The MPT has also been recommended for red cell antibody screening, provided that appropriate positive and negative controls are used (Ferrer et al. 1985); the test carried out in microplates was found to be as sensitive as the tube test (Lown and Ivy 1988).
False-Positive Results in Agglutination Tests
The commonest cause is rouleaux formation. Dilution of serum with an equal volume of saline greatly diminishes or completely abolishes rouleaux. Dextran is a potential source of trouble but only high-molecular-weight preparations (no longer used as plasma substitutes) cause rouleaux formation of untreated red cells. Enzyme-treated red cells may be intensely aggregated even by Dextran 40 (Selwyn et al. 1968).
The next commonest cause of false-positive results is the presence of cold autoagglutinins. These antibodies (usually anti-I) produce strong agglutination up to a temperature of about 25°C, or even sometimes up to 32°C, but are very seldom active at a temperature of 37°C. If red cells and serum are not pre-warmed to 37°C before being mixed, subsequent warming to 37°C may fail to disperse agglutination completely.
When there is difficulty in distinguishing between rouleaux formation and autoagglutination it is helpful to prepare dilutions of the serum in saline (1 in 2 and 1 in 4) and to incubate these with the subject’s own red cells, first at 4°C and then at 37°C. Potent autoagglutinins should react very strongly at 4°C and weakly or not at all at 37°C; furthermore, diluting the serum 1 in 4 should have little or no effect on the degree of agglutination. By contrast, rouleaux formation should be more pronounced at 37°C than at 4°C; it should be very much weaker in a 1 : 2 dilution than in undiluted serum and should be completely absent in the 1 : 4 dilution.
Spontaneous Agglutination of Antibody-Coated Cells in Albumin
A problem that arises when albumin is used as a potentiator of agglutination is that red cells sensitized with an autoantibody in vitro or in vivo may agglutinate spontaneously in concentrations of albumin as low as 6% (Garratty et al. 1984). It is therefore essential, as a routine, to carry out a control test in which the red cells are mixed with potentiating agent alone. The term potentiating agent is used because some reagents which contain albumin may contain other potentiating agents so that the control used must be one supplied by the manufacturer of the reagent containing everything except the reagent antibody (White et al. 1974; Garratty et al. 1984).
Mixed Field Agglutination
This term is used to describe the presence of agglutinated and unagglutinated cells in a red cell suspension treated with an agglutinin. The term is often used to imply that two phenotypically distinct populations of red cells are present, as in mosaics, chimeras, subjects who have been transfused, women whose circulation contains fetal red cells, and subjects some of whose red cells have undergone T or Tn transformation. On the other hand, a similar appearance may be seen in red cell suspensions of a single phenotype, either when the red cells have relatively few antigen sites (e.g. ‘weak’ varieties of A) or when the agglutinin is of low titre. Lutheran antibodies characteristically produce a mixed field appearance of large agglutinates with many free cells and anti-Sda produces small agglutinates in a sea of free cells.
Although mixed field agglutination is included in the present section under the general heading of ‘agglutination of red cells suspended in saline’, it may be observed in any agglutination reaction, for example between antibody-coated red cells and antiglobulin serum. For example, when the direct antiglobulin test (DAT) is performed in a patient who is having a DHTR, a mixed field may be observed, for (as a rule) only the donor cells are agglutinated.
Again, if an IAT with anti-D is performed on red cells from a D-negative mother whose circulation contains a substantial number of D-positive red cells, owing to a transplacental haemorrhage (TPH) from her fetus, a mixed field will be observed and may be misinterpreted as meaning that the mother has a weak D antigen. When a mixed field due to the presence of two phenotypically distinct populations is suspected, there are many tests that can be done to confirm or refute the suspicion. For example, tests may be made with sera containing alloantibodies of various specificities, which may reveal that there are two populations of red cells present, differing with respect to many antigens. Alternatively, the agglutinated cells may be separated from the agglutinated cells by differential sedimentation and the agglutinated cells may then be disagglutinated and tested.
In detecting the presence of two different populations of red cells, the gel test (see ‘The gel test’) is excellent. In both techniques, one population of cells will remain at the top of the gel (agglutinated) and the other will go to the bottom of the tube or column (unagglutinated). The gel test has been successfully used to detect mixed red cell populations in patients who had received a bone marrow transplant (Zago-Novaretti et al. 1993).
Tests for Lysis of Red Cells
Tests for alloantibodies that lyse red cells (haemolysins) are no longer used in blood grouping: first, because, apart from some examples of anti-A and anti-B, most blood group antibodies will not readily lyse red cells and, second, because the lytic property of serum deteriorates rapidly on storage due to the decay of complement. Nevertheless, in some circumstances, it is valuable to know whether or not a serum sample is lytic: (1) if a serum causes specific lysis, it establishes the fact that it contains a complement-binding antibody; (2) if a serum is readily lytic in vitro at 37°C, the antibody is likely to cause intravascular lysis of incompatible red cells (although this information would usually be of interest only in a retrospective investigation); (3) in circumstances in which group O blood has to be used for transfusion to group A or B subjects, a screening test for lytic anti-A and anti-B may be the best for detecting really ‘dangerous’ group O donors (see Chapter 4); and (4) if an infant is suspected of having ABO haemolytic disease, the diagnosis is virtually excluded by showing that the mother’s serum will not lyse the infant’s red cells (Crawford et al. 1953).
Apart from anti-A and anti-B, the commonest lytic antibody encountered is anti-Lea. Other strongly lytic alloantibodies are all rare, i.e. anti-H (in hh–subjects), anti-P, anti-PP1Pk and anti-Vel. Autohaemolysins are described in Chapter 7.
In crossmatching tests, lytic antibodies may be a cause of false-negative results; most of the cells may be lysed and the remainder may be unagglutinated. It is therefore essential to examine the supernatant for lysis before examining the cell button for agglutination, and before washing the cells in the antiglobulin test.
Use of Human Serum As a Source of Complement
In testing for haemolysins, it is preferable to use very fresh serum, i.e. serum taken within the previous 2 h (see Chapter 4). When stored serum is tested for haemolysins, fresh serum should be added as a source of complement.
In testing for anti-A or anti-B lysins, group O serum from which anti-A and anti-B have been absorbed may be used. In detecting lytic anti-Lea or anti-Leb, serum from Le(a– b–) subjects, free from Lewis antibodies, may be used. Alternatively in detecting lysis by Lewis antibodies, a two-stage test may be used and then the Lewis group of the serum used as a source of complement in the second stage is of no importance (Polley and Mollison 1961).
Use of Animal Serum As a Source of Complement
For details, see Chapter 3.
Importance of Ratio of Serum to Cells
For details, see Chapter 4.
Enzyme Treatment of Cells
Some examples of anti-Lea, anti-Leb and anti-Jka will lyse untreated red cells; other examples will lyse only enzyme-treated cells.
51Cr Release Method
The technique of estimating the release of 51Cr from 51Cr-labelled group A1 red cells incubated with anti-A is referred to in Chapter 9. In addition to being convenient for demonstrating very weak haemolysins this method is useful for demonstrating the presence of a very small proportion of A1 red cells in a mixture of A1 and O cells, for example in measuring the proportion of A1 cells in blood group chimeras (Booth et al. 1957). The method can also be used to estimate the survival of transfused A cells in an O subject (see Chapter 11 and Appendix 7).
The Antiglobulin Test (Coombs’ Test)
By definition, non-agglutinating (incomplete) antibodies are those that fail to agglutinate red cells suspended in saline. In the antiglobulin reaction, red cells coated with incomplete antibodies, for example IgG anti-Rh, are agglutinated by anti-IgG, which links the IgG molecules on neighbouring red cells. The principle of the test was described by Moreschi (1908) who showed that if rabbit red cells were incubated with a dose of goat anti-rabbit red cell serum too small to produce agglutination, and then washed, they were strongly agglutinated by rabbit anti-goat serum.
The antiglobulin test was rediscovered and introduced into clinical medicine by Coombs and co-workers (1945) who showed that it could be used either to detect incomplete blood group antibodies in serum – the IAT – or to detect the sensitization of red cells in vivo, as in haemolytic disease of the newborn (Coombs et al. 1946) – the DAT.
Most non-agglutinating blood group antibodies are IgG and are detected by an anti-IgG serum. A few IgM antibodies are incomplete, for example Lewis antibodies, which, at 37°C, usually fail to agglutinate red cells but may be detectable by the IAT using anti-IgM (Polley et al. 1962), although they are best detected with anti-complement.
The fact that an anti-human globulin (AHG) reagent might react with complement components on red cells was first described by Dacie and co-workers (1957) and it was subsequently shown that the main complement components detected were C4 (Jenkins et al. 1960; Pondman et al. 1960) and C3 (Harboe et al. 1963).
Antibodies Required in Antiglobulin Reagents
As described above, AHG reagents may be used to detect IgM and IgA, as well as IgG antibodies on red cells and may also be used to detect various components of complement.
In considering which antibodies are required in AHG reagents, a distinction must be made between reagents used in detecting alloantibodies by the IAT and those used in diagnosing sensitization in vivo by the DAT.
Antiglobulins for Indirect Tests
In the detection of alloantibodies, using the routine spin-antiglobulin test, anti-IgG is clearly essential but anti-IgM is not required. All incomplete IgM antibodies described so far bind complement and can be detected more readily with anti-complement than with anti-IgM, because each bound IgM molecule leads to the binding of many complement molecules. Some blood group antibodies (e.g. anti-A, anti-B, anti-D) may be partly IgA (Adinolfi et al. 1966) but are then always also partly IgG so that anti-IgG can be used for their detection. A single weak example of anti-K was wholly IgA (Pereira et al. 1989).
In detecting alloantibodies by conventional antiglobulin techniques the presence of anti-complement in the AHG reagent often leads to stronger reactions and very occasionally leads to the detection of antibodies, which would otherwise be missed (for references, see the eighth and earlier editions of this book). When using the conventional spin-antiglobulin test for the detection of alloantibodies the reagent must contain anti-complement. On the other hand, reagents lacking anti-complement are often used in the PEG–IAT, and in the MPT followed by an IAT. Reagents containing anti-complement as well as anti-IgG can be used in these tests but particular care must then be taken to avoid false-positive results.
In deciding which anti-complement components to include in AHG reagents, there are good grounds for preferring anti-C3 to anti-C4: in the activation of complement by the classical pathway, more C3b than C4b is bound, and during storage at 4°C, more C4d than C3d is bound to red cells non-specifically (Engelfriet 1976; Garratty and Petz 1976). The choice of the particular anti-C3 component is more difficult. When C3b is bound to red cells it is converted rapidly to iC3b, which reacts with anti-C3c, -C3d and -C3g. iC3b is then further cleaved; C3c is split off, leaving only C3d and C3g on the cell surface (see Chapter 3). The advantage of using anti-C3c is that C3c is found on cells only as a consequence of antibody binding, although the use of too high a concentration of anti-C3c may give false-positive results (Voak et al. 1986a). The disadvantage of using anti-C3c is that the period of incubation in the IAT must be limited to about 30 min, after which the amount of C3c remaining on the cells may be suboptimal. As some laboratories use longer periods of incubation, it has been recommended that either anti-C3d (Giles and Engelfriet 1980) or anti-C3g (Voak et al. 1986a) should be included in AHG reagents. In both cases, the concentration must be too low to detect the small amount of C3dg present in small amounts on normal red cells and in larger amounts on stored cells and on cells incubated in fresh normal serum (see Stratton and Rawlinson 1976). When using polyclonal anti-C3d, false-positive results are not due solely to the detection of C3d but may be due to the synergistic effect of various antibodies against complement components, each present on the red cells in too low a concentration to be detected separately. Unwanted positive results are less of a problem when using monoclonal anti-C3d, although even then there may be trouble (Voak et al. 1986a).
One of the two polyspecific antiglobulin reference reagents, made available by the ISBT and the ICSH, reagent RIIIM, is a blend of polyclonal anti-IgG and monoclonal anti-C3c and anti-C3d. The other reference reagent (R3P) is entirely polyclonal (see Engelfriet and Voak 1987). Antiglobulin reagents that are blends of murine monoclonal anti-IgG and either monoclonal anti-C3d or monoclonal anti-C3d and C3c have been described and found to be as satisfactory as reagents containing polyclonal anti-IgG (Betremieux et al. 1994; Moulds et al. 1994). In 1999 the ISBT/ISCH working party on international reference reagents produced a third reference reagent for antihuman globulin to replace those made available in 1987 (Case et al. 1999).
AHG Reagents for Direct Tests
In the detection of autoimmune haemolytic anaemia (AIHA) and drug-induced haemolytic anaemia, using the DAT, anti-IgM is not needed in addition to anti-IgG because IgM autoantibodies are always complement binding and can be detected better with anti-complement. Anti-IgA is needed very rarely because it is very rare for IgA alone to be detectable on the red cells (see Chapter 7). It is simpler to use monospecific anti-IgA to test cells that are negative with a polyspecific reagent than to include anti-IgA in the polyspecific reagent.
The main components of complement found on red cells from patients with AIHA are C3dg and C4d (see Chapter 7). So far, no case of AIHA has been described in which one of these components has been present without the other. Because polyspecific reagents for the IAT contain anti-C3d or anti-C3g, which, although present in restricted amount, will readily detect the large amounts of C3dg found on red cells of patients with AIHA, the same polyspecific reagent can be used for the DAT and the IAT.
Production of Antiglobulin Reagents
In most laboratories, commercially available AHG reagents are used. For those who wish to produce their own reagents, a survey of methods of production is available (Engelfriet et al. 1984).
Heteroagglutinins in Antiglobulin Reagents
When AHG reagents produced in animals are used, heteroagglutinins are a potential source of trouble; methods of dealing with this problem have been described previously (seventh edition, pp. 507–508). If AHG reagents are to be used with enzyme-treated cells, residual heteroagglutinins must be removed by absorption with enzyme-treated cells.
The Reaction with Bound Immunoglobulin
In the IAT, red cells are first incubated with serum to allow the uptake of antibody (and, in some cases, the binding of complement) and are then washed and tested with an AHG reagent.
Reaction between Anti-IgG and IgG-Coated Red Cells
Using radioiodine-labelled antiglobulin serum, the maximum number of anti-IgG molecules that can combine with an anti-D (IgG) molecule on a red cell surface was estimated to be six to nine by Costea and co-workers (1962) and Rochna and Hughes-Jones (1965). Near-saturation of the antigen sites of an IgG (anti-D) molecule is obtained by having a free equilibrium concentration of 10–15 µg of anti-IgG per ml; that is to say, after the uptake by anti-D of the maximum number of anti-IgG molecules, 10–15 µg of anti-IgG per ml should remain in solution. With an initial IgG concentration of 15 µg/ml the amount of antibody taken up by sensitized red cells after 4 min is 95% of the final equilibrium value (Rochna and Hughes-Jones 1965).
Prozones
If an excess of anti-IgG serum is added to a sample of IgG-sensitized red cells, agglutination is inhibited. This prozone phenomenon was investigated by van Loghem and co-workers (1950) and considered to be due to the fact that, with antiglobulin in excess, all the IgG molecules attached to the red cells are coated with anti-IgG so that no ‘bridges’ can be formed by particular anti-IgG molecules reacting with unsaturated IgG molecules on different red cells. An example of a prozone is shown in Table 8.2.
Table 8.2 Comparison of optimal dilutions of a particular anti-human globulin serum (Goat, H39), for cells sensitized with an example of anti-Rh (Avg.) and one of anti-Jka (Cro.).
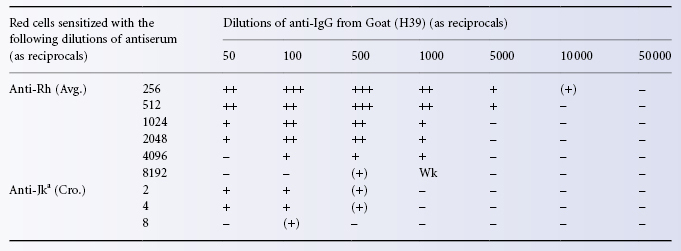
This type of prozone can be eliminated by washing the cells after incubation with AHG; presumably removal of excess unbound AHG facilitates lattice formation by removing competition between bound and unbound AHG molecules (Salama and Mueller-Eckhardt 1982).
Optimal Concentration of Anti-IgG
Surprisingly, there is no apparent correlation between the anti-Ig concentration and the serological activity of anti-IgG reagents (Gardner et al. 1983). Accordingly, such reagents must be assessed serologically; see Engelfriet and Voak (1987).
A single dilution of a particular AHG reagent is usually optimal for detecting almost all examples of anti-D. On the other hand, a lower dilution may be optimal for detecting occasional alloantibodies of other specificities. There is some evidence that aberrant behaviour with anti-IgG sera is related to the specificity of the alloantibody concerned. Specificities which are suspect in this regard are anti-Jka (see Table 8.2), anti-Fya (Pollack et al. 1962) and anti-S, -s, -Xga, -Yta and -Vel (Issitt 1977). It should be emphasized that many alloantibodies of these specificities are best detected with the dilution of AHG reagent that is optimal for detecting anti-D but it may be necessary to compromise by selection of a somewhat lower dilution for routine use.
Because of the heterogeneity of Ig molecules the use of many donors to provide pooled IgG, and the pooling of anti-IgG from many immunized animals, is desirable in producing reagents for routine use.
Diluent for Antiglobulin Reagents
AHG reagents are usually provided at their optimal dilution and for storage at 4°C. Various diluents are employed; the following one is convenient: 0.1 mol/l of NaCl, 0.05 mol/l of phosphate pH 7.2, 1 g/l of Na4 EDTA 2H2O, 10 g/l BSA and 1 g/l of sodium azide. Use of a low-ionic-strength diluent does not appear to enhance reactions of AHG reagents, perhaps because bound IgG antibodies extend beyond the ‘ionic cloud’ surrounding red cells (Leikola and Perkins 1980a). However, the use of low-ionic-strength antiglobulin serum in the LISS–IAT has been found to increase the sensitivity of the test (Ahn et al. 1987).
Dyes may be added to AHG reagents to provide a means of checking that the reagent has been added to tubes. Green is the colour most frequently used for commercial polyspecific reagents and a suitable depth of colour is provided by mixing 0.08 g/l of Ariavit tartrazine (E102) with 0.02 g/l of Patent blue V (E131).
Relationship between Number of Bound IgG Molecules and Reactions with Anti-IgG
Using the spin-antiglobulin test, the minimum number of IgG anti-D molecules per cell detectable with anti-IgG is between 100 and 150 (Romano et al. 1973; Burkart et al. 1974; Stratton et al. 1983). The minimum detectable number of IgG anti-A and anti-B molecules per red cell bound to the red cells of either adults or newborn infants is also about 150 (Romano et al. 1973). In normal subjects with a negative DAT the number of IgG molecules per red cell was found to be in the range of 5–90 (Merry et al. 1982). The findings of Jeje and co-workers (1984) were almost identical. In both series the average number of IgG molecules per red cell was between 30 and 40.
A correlation exists between agglutination strength and the number of IgG molecules bound per cell in both the DAT and the IAT (Merry et al. 1984); in the IAT anti-K bound under low-ionic-strength conditions required a greater number of bound molecules for a given agglutination strength than antibodies of other specificities. The number of molecules per cell required for maximal agglutination with anti-IgG is in the range of 500–2000 (Petz and Garratty 1980; Schmitz et al. 1981; Merry et al. 1984). As the number of IgG molecules bound per red cell in AIHA frequently exceeds 2000, the strength of agglutination in the DAT is of limited value as an indicator of the degree of antibody sensitization.
Inhibition of Anti-IgG by IgG in Solution
When using an AHG reagent containing about 10 µg of anti-IgG per millilitre, obvious weakening of the reaction between anti-IgG and IgG-coated cells is not likely to occur unless the level of IgG in the suspending medium reaches about 10 µg/ml. As normal serum contains about 10 µg of IgG per millilitre it must be diluted at least 1000 times during washing of antibody-coated red cells to avoid false-negative results. A better safety margin is a dilution of about 5000.
Suppose that one incubates 0.08 ml (two drops) of test serum with 0.04 ml of cell suspension, that 4 ml of saline is used for each wash and that 0.1 ml of supernatant is left with the cells after each wash: a single wash then dilutes the serum only 18-fold; three washes will dilute the original serum almost 6000-fold and will therefore be adequate. Nevertheless, as many commercial AHG reagents appear to contain much less than 10 µg of IgG/ml, it is advisable to wash the red cells four times. The danger of false-negative reactions owing to inadequate washing is reduced when two volumes of AHG reagent are used to one volume of washed cell suspension. When cell-washing centrifuges are used, correct maintenance of the machines is essential (Voak et al. 1986b).
Reactions of Anti-IgM
Many IgM antibodies are agglutinins active at 37°C and the reactions of these antibodies are not enhanced by the addition of anti-IgM. However, certain IgM antibodies act as agglutinins only at temperatures up to about 25°C or 30°C, and at 37°C may act as incomplete antibodies and then be demonstrable by the antiglobulin test using anti-IgM. Polley and co-workers (1962) found that the following antibodies behaved in this way: anti-Lea (eight out of eight examples), anti-HI and anti-P1 (single, selected potent examples) and allo-anti-I (from an i donor). In addition to these cold antibodies, a few examples of warm incomplete IgM antibodies were found that were capable of sensitizing red cells to agglutination by anti-IgM, namely one out of three examples of anti-K and 3 out of 15 examples of anti-Jka (Polley et al. 1962).
The agglutination produced by anti-IgM is weak compared with that produced by anti-IgG, perhaps because of the small number of IgM molecules attached to the red cells. Therefore, when the antibody concerned binds complement it is always easier to detect it by incubating red cells with antibody and complement and then testing the cells with an anti-complement reagent.
Red cells coated only with IgM can be prepared by sensitizing Le(a+) cells with serial dilutions of EDTA-treated serum containing potent anti-Lea. At some dilution the anti-Lea fails to agglutinate the cells but sensitizes them to agglutination by anti-IgM. Another method which has been used successfully is to prepare purified IgM anti-D and treat it with 2-mercaptoethanol (2-ME). The treated preparation will fail to agglutinate D-positive red cells but will sensitize them to agglutination by anti-IgM. Anti-Lea treated with 2-ME will not sensitize Le(a+) red cells to agglutination by anti-IgM because the IgM subunits have a very low affinity (Holburn 1973).
Reactions of Anti-IgA
Anti-A and anti-B are quite commonly partly IgA, as are potent examples of anti-D; Lutheran antibodies may also be partly IgA. Relatively few tests have been made to see whether antibodies of other specificities, for example anti-K, are also partly IgA.
Red cell antibodies are seldom made solely of IgA; although a few autoantibodies of this kind have been reported, only a single such alloantibody has been described (see Chapter 3).
A convenient way of preparing red cells coated with IgA (and also IgG) is simply to take a number of anti-D sera (e.g. 10) from hyperimmunized subjects and use them to sensitize D-positive red cells. Several samples in the batch are likely to sensitize red cells to anti-IgA. The fact that the reactions are not due to contaminating anti-IgG can be confirmed by adding IgG (0.1 g/l) to the anti-IgA serum. An alternative method is to use chromic chloride to couple IgA myeloma protein to red cells (see Chapter 13).
Anti-IgA sera, like anti-IgG, exhibit prozones and usually react optimally at a considerable dilution, such as 1 in 500 (seventh edition, p. 514).
The Reaction with Bound Complement
Unlike anti-IgG reagents, anti-complement reagents produced in animals do not exhibit obvious prozones. On the other hand, monoclonal anti-C3d reagents, because of their high antibody concentrations, exhibit prominent prozones so that, unless the reagents are adequately diluted, negative reactions may be observed (D Brazier, personal communication, 1985).
When testing red cells coated with complement components, less washing of the coated cells is required after incubation with serum because of the relatively low concentrations of C3 and C4 in serum (see seventh edition, p. 514). Anti-C3g is not neutralized by serum at all because C3g is not expressed on native C3.
Preparation of Complement-Coated Red Cells
For methods, see Engelfriet et al. (1987).
Quantification of Anti-Complement Components
The optimal concentrations of anti-C3b and anti-C3c are 1–2 µg/ml, i.e. similar to those of anti-IgG (Gardner et al. 1983). In the ISBT/ICSH reference antiglobulin reagents, the titre of anti-C3c with red cells coated with iC3b is 32–64; that of anti-C3d with C3d-coated cells is 2–4 and should not be higher if false-positive results are to be avoided.
Technique of Antiglobulin Tests
In the IAT, red cells are first incubated with serum to allow the uptake of antibody and, in some cases, complement, and are then washed and tested with an AHG reagent. In the DAT the cells are simply washed and tested. The uptake of antibody and complement in the indirect test will be discussed first.
The Uptake of Antibody
The sensitivity of antibody detection is affected by several variables which determine the rate and extent of antibody uptake.
Effect of the Ratio of Serum to Red Cells
The amount of antibody taken up per red cell is at a maximum when the ratio of serum to cells is about 1000 : 1 (Hughes-Jones et al. 1964b). Under normal conditions the ratio is much lower. For example, when one volume of serum is incubated with one volume of a 3% suspension of red cells, the ratio of serum to cells is 33 : 1. The effect of lowering the cell concentration from 2.5% to 0.5% is shown in Table 8.3.
Table 8.3 Effect of: (1) red cell concentration (2.5% vs. 0.5%) and (2) saline vs. LISS as a suspending medium, on the reactions of Fy (a +) cells with anti-Fya in the indirect antiglobulin test.
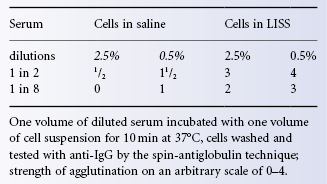
Dropper pipettes used for dispensing serum and commercially supplied red cell suspensions were found to deliver between 17 and 43 drops/ml. Moreover, the PCV of the red cell suspensions varied by a factor of 2. When two drops of serum were added to one drop of cell suspension, the ratio of serum to cells varied from 19 : 1 to 70 : 1 (Beattie 1980).
The titre in the normal-ionic-strength solution (NISS) IAT of most Rh antibodies and of some Kell antibodies was four times higher when four volumes of serum were used instead of one volume with 3% cell suspension, i.e. a serum–cells ratio of about 130 : 1. Similar results were obtained in tests with ficin-treated cells and in low-ionic polycation tests (Ahn et al. 1987).
Effect of Period of Incubation
The time taken for the maximum uptake of antibody depends on the concentrations of antigen and antibody and also on the binding constant of the reaction. Typical figures for incubation at 37°C, with a final concentration of DCcee cells of 2% in ‘NISS’ and of anti-D, with a binding constant of 108 l/mol, of 1 µg/ml are: maximum uptake after 4 h; 40% of this amount after 15 min, 87% after 1 h and 99% after 2 h (NC Hughes-Jones, personal communication; see also Hughes-Jones et al. 1962). Evidently, the period for which cells and serum are incubated in serological tests is somewhat arbitrary; 30 min is adequate to detect most antibodies, although some weak antibodies need longer incubation (see AABB, 1993); in cases of urgency, shorter periods may be used.
Effect of Temperature
In detecting Rh antibodies, incubation at 37°C is optimal (see Chapter 3). In IATs, scores were never higher at 30°C than at 37°C, not only with Rh antibodies but also with those of the Kell, Duffy and Kidd systems. On the other hand, although incubation at 22°C gave lower scores with about 50% of Rh antibodies, it gave the same scores with Kidd antibodies and with 80–90% of Duffy and Kell antibodies. At 10°C scores were as high as at 37°C with 60% of SsU antibodies (Arndt and Garratty 1988). Many monoclonal Rh antibodies react optimally at room temperature.
Effect of Low Ionic Strength
As discussed above, the uptake of antibody is much more rapid at low ionic strength. With many alloantibodies, the titre was found to be increased when red cells were suspended in low-ionic-strength medium but the effect on undiluted serum was not tested (Elliot et al. 1964; Hughes-Jones et al. 1964b). In view of the observation that red cells exposed to serum at low ionic strength take up complement non-specifically, there was, for a time, reluctance to use a low-ionic-strength medium for routine tests. However, Löw and Messeter (1974) showed that when red cells were suspended in a solution of sodium glycinate containing 0.03 mol/l NaCl, false-positive results were not a problem, and this finding was confirmed (Moore and Mollison 1976). The latter authors gave details of a more convenient method of preparing LISS; incidentally, LISS is prone to growth of bacteria and is best sterilized by filtration. A method of performing IATs using LISS-suspended red cells is as follows.
Red cells are washed twice in saline and then once in LISS; a 3% suspension of red cells in LISS is then prepared. One volume of this suspension is added to an equal volume of serum (drops may be used except with plastic test tubes, for which it is better to use some form of automatic pipette to ensure that the drops of cell suspension and serum are equal in volume). The mixture is incubated at 37°C for 10 min and the red cells are then washed three times; use of LISS for cell washing leads to only slightly stronger reactions than when the cells are washed in saline. Finally, the washed cells are tested with antiglobulin serum in the usual way.
In the technique of Löw and Messeter (1974), ionic strength is reduced by about 20%. In the polybrene technique described by Lalezari and Jiang (1980), ionic strength is reduced by about 80% and the period of incubation, at room temperature, is reduced to 1 min (see ‘Role of ionic strength’); there is a similar reduction in ionic strength and incubation period in the test of Szymanski and Gandhi (1983).
The chief advantage of suspending red cells in LISS rather than in saline lies in the increased rate of uptake of antibody. Although maximum antibody coating may be observed after a period of incubation as short as 5 min, it has been recommended that 10 min of incubation should be used as a routine because results are sometimes stronger at 10 min than at 5 min (Moore and Mollison 1976).
The only clinically significant antibody that tends to react less well in a low-ionic-strength medium is anti-K. In one investigation, 3 out of 16 samples failed to react, and one of these was associated with an HTR (Molthan and Strohm 1981). In another investigation, 2 out of 195 examples were detected with LISS but not with saline, and the remainder was detected with both (Dankbar et al. 1986). The problem of failing to detect anti-K at low ionic strength can be overcome by using a serum–cell ratio of at least 40 : 1 (Voak et al. 1982).
Using 125I-labelled anti-IgG, Merry and co-workers (1984) observed an accelerated rate of uptake in LISS of antibodies of several specificities but anti-K antibodies were the exception, less antibody being bound in LISS than in saline.
A disadvantage of the use of LISS is that occasional serum samples give a positive IAT with all red cells, including the subject’s own cells (see, for example, Morel and Vengelen-Tyler 1979). Red cells suspended in LISS give enhanced reactions with common cold alloantibodies such as anti-A1 and anti-P1, but this potential disadvantage can be almost completely overcome by eliminating a room temperature incubation phase and by warming red cells and serum to 37°C before mixing them.
The use of a low-ionic-strength antiglobulin serum in the LISS–IAT was found to increase the sensitivity of the test and, particularly with stored sera, to result in fewer non-specific reactions than with the conventional LISS–IAT (Ahn et al. 1987).
Use of Albumin (BSA–IAT)
Although albumin enhances the uptake of some alloantibodies, its use adds to the complexity and cost of testing. As emphasized above, the simplest and most effective way of increasing sensitivity is to increase the ratio of serum to cells.
‘Spin-Tube’ Antiglobulin Test
In this test, a weak (3%) suspension of washed sensitized red cells is mixed with antiglobulin serum in a tube, which is then briefly centrifuged, so as to produce a cell button without packing the cells too firmly. The cells are gently resuspended and examined for agglutination either with a hand lens or under the low power of a microscope.
Use of an inverted microscope provides a convenient means of examining the cells microscopically within the tube and avoids the need to transfer the cells to a microscope slide.
Although an immediate spin after adding red cells to antiglobulin serum seems to be optimal for detecting IgG-coated red cells, red cells coated with IgA may be better detected after cells and antiglobulin serum have been in contact for a longer period, for example 5–10 min (Sturgeon et al. 1979). When the results after an immediate spin are negative, therefore, the tube may be respun after 10 min and read again. C3d reactions may be stronger when cells and antiglobulin serum are incubated before being centrifuged (G Garratty, personal communication).
Use of Enzyme-Treated Cells
Some antibodies that fail to sensitize untreated red cells to agglutination by antiglobulin serum may be detected if enzyme-treated red cells are used (Unger 1951). The enzyme–antiglobulin method is particularly suitable for detecting anti-Jka (van der Hart and van Loghem 1953). It will also reveal the presence of the antigen Lea on red cells, which, by other methods, appear to be O Le(a– b+) (Cutbush et al. 1956).
As described above, false-positive results obtained with enzyme-treated cells are sometimes due to the presence of heteroagglutinins in antiglobulin sera. Another potential source of false positives is overtreatment with the enzyme.
Polyethylene Glycol Indirect Antiglobulin Test (PEG–IAT)
PEG, a water-soluble polymer, potentiates red cell–antibody interactions in the antiglobulin test (Nance and Garratty 1987). A 20% solution of PEG of molecular weight 4000 was found to be optimal. Of 25 weak antibodies tested, 64% reacted more strongly in the PEG–IAT than in LISS–IAT or in the MPT, 28% reacted equally well in all three techniques and 8% reacted more weakly in the PEG–IAT test. It was found that many false-positive reactions occurred if the antiglobulin serum used contained anti-complement.
Several studies have confirmed the superiority of the PEG–IAT over the LISS–IAT and the BSA–IAT. Reactions tend to be stronger in the PEG–IAT than in the other two tests and a few clinically significant antibodies are detected only in the PEG–IAT and not in the BSA–IAT or the LISS–IAT (Slater et al. 1989; De Man and Overbeeke 1990; Wenz et al. 1990; Shirey et al. 1994; Barrett et al. 1995).
In detecting most warm antibodies, the PEG–IAT, in which anti-IgG or -Ig is used, is more sensitive than the BSA–IAT, although in this latter test a polyspecific reagent containing anti-complement is used. On the other hand, a few antibodies were detected in the BSA–IAT or the LISS–IAT but not in the PEG–IAT: an anti-Jka (Slater et al. 1989); two anti-Vel and one anti-P, ascribed to low affinity of the antibodies (MAM Overbeeke, personal communication); and Lutheran antibodies, whose reactivity seems to be reduced by PEG (Fisher 1990).
As mentioned above, the MPT, particularly if it is followed by a test with antiglobulin serum (MPT–IAT), is also more sensitive than the conventional antiglobulin test. In fact it is as sensitive as the PEG–IAT (R Knight, personal communication). For the antiglobulin test following the MPT, an antiglobulin reagent without anti-complement is used. If either the MPT–IAT or the PEG–IAT replaces the conventional antiglobulin test for the detection of alloantibodies, anti-complement will no longer be required in the antiglobulin reagent.
The Gel Test
In this test, special microtubes filled with Sephadex gel are used (Lapierre et al. 1990). For the detection of saline agglutinins a neutral gel is applied, and for the antiglobulin test, a gel containing antiglobulin reagent. Gels containing red cell antiserum can also be used. The red cells are centrifuged through the gel. In a negative reaction, all cells collect at the bottom of the tube, whereas in a positive reaction the cells are trapped in the gel. The test is easy, sensitive and reproducible. Advantages are that the antiglobulin test can be performed without washing the cells, that small quantities of reagents are used and that after the reaction has occurred the gels can be kept for at least 24 h, allowing second opinions to be sought and photocopies of the tubes to be made.
With the commercial version of the test (DiaMedID Micro Typing System) contradictory results were at first reported. Some found it to be more sensitive than other tests, such as the MPT, a microplate IAT, the LISS–IAT and a two-stage papain test (Bromilow et al. 1991). Others found it less sensitive than a semi-solid-phase microplate IAT (Voak et al. 1991) or the LISS–IAT (Phillips 1992) and even slightly less sensitive than the saline-IAT (Pinkerton et al. 1993a,b). However, in most subsequent studies the gel test has been found to be more sensitive than the traditional antiglobulin test (Bromilow et al. 1993; Phillips and Whitton 1993; Lillevang et al. 1994; Titlestad et al. 1997). At the same time, the DiaMed-ID test has been found to be less reliable than the immediate spin-tube technique in detecting ABO incompatibility between serum from B donors and A2B cells (Cummins and Downham 1994). Phillips and co-workers (1997) proposed that the failure of the gel test to detect weak ABO incompatibilities results from shear forces applying during centrifugation through the gel, which do not occur in the spin-tube method. They further note that this failure of the gel method is unlikely to have any clinical significance because the weak ABO antibodies missed react poorly, if at all, at 37°C. Performing the DAT with the gel test is easier and more sensitive than spin-tube and has the added advantage of minimizing exposure of blood bank personnel to risk from infectious samples, particularly in areas of the world where blood-borne diseases are prevalent (Nathalang et al. 1997). A variation of the gel test in which glass beads are placed in a microcolumn together with a diluent has also been developed (Reis et al. 1993).
Tests in Microplates
An antiglobulin test in microplates was first described by Wegmann and Smithies (1966) and found to be suitable for routine use (Crawford et al. 1970). The reliability of the antiglobulin test in microplates for the detection of red cell alloantibodies was confirmed in three large studies (Crawford et al. 1988). Because of the small volume in which the reaction is carried out, the cells should be washed four times (Crawford et al. 1988). Several variations of this technique are used (Gordon and Ross 1987). Both V- and U-shaped wells can be used and to reduce stickiness the use of saline containing 0.1% BSA and/or 0.02% Tween 20 for washing the cells is recommended. The advantages of the test are sensitivity and the need for only very small quantities of reagents and cells.
Antiglobulin reagents standardized for spin-tube tests may produce a prozone in liquid-phase microplate tests. Accordingly, the optimal dilution of each batch of antiglobulin reagent must be determined (Voak et al. 1990). Dilution may compromise anti-complement activity and make anti-IgG more prone to neutralization. Washing procedures, particularly when automated, must be strictly controlled.
The Detection of Bound Complement
When fresh serum is available and it is not anticomplementary, optimal results are obtained by carrying out the test in exactly the same way as for detecting bound IgG. When testing stored serum in which the complement components may have decayed, fresh normal serum should be added. For example, one volume of fresh normal serum may be added to three volumes of the antibody-containing serum, and the IAT then carried out on the mixture.
Lewis antibodies in the test serum may inadvertently be neutralized by the addition of Lewis substances to the serum, unless Le(a– b–) serum is used as a source of complement. On the other hand when a two-stage test is used, the red cells being incubated first with antibody and then with complement, fresh serum of any Lewis group may be used as a source of complement (Polley and Mollison 1961).
Two-Stage Test
Stored sera may become strongly anticomplementary. It may then become difficult to detect the presence of antibodies that are best detected (such as anti-Lea) by a technique based on complement binding. The difficulty can be overcome by first incubating the red cells with EDTA-treated serum, washing them and then incubating them with fresh serum. The presence of EDTA in the first stage prevents the binding of altered complement (once described as ‘complementoid’), which may otherwise block the binding of normal complement in the second stage of the test (Polley and Mollison 1961).
False-Negative Results in Antiglobulin Tests
The most important cause of false-negative results is failure to wash the red cells adequately. To demonstrate that a negative result is not due to neutralization of antiglobulin by residual serum, a drop of D-sensitized red cells should be added. In tube tests, the mixture is spun before being read. An inverted microscope is particularly convenient for this procedure, as the antiglobulin is all retained within the tube. It is particularly important that the D-sensitized red cells are not too strongly sensitized, because strongly sensitized cells may be agglutinated by partially neutralized AHG reagent. The cells should not give stronger reactions than 1+ to 2+ in the IAT (Voak et al. 1986b).
Another important cause of false-negative reactions is excessive agitation in reading the test (Voak et al. 1986b). The ‘tip and roll’ procedure, combined with reading under an inverted microscope, or careful transfer of the cell button to a slide with reading under a standard microscope are advised.
Wash solutions should be buffered to pH 7.0–7.4. Failure to detect clinically significant antibodies may be due to the use of wash solutions with pH values below pH 5 or of solutions that have been autoclaved and stored in plastic containers (Bruce et al. 1986).
False-Positive Results in Antiglobulin Tests
One may distinguish between genuine false-positive results, due to unwanted antibodies such as heteroagglutinins in the antiglobulin serum (see ‘Heteroagglutinin in antiglobulin reagents’), and those positive results that are ‘true’ in that they indicate the presence of globulin on the red cells, but are unwanted, in that they have no obvious clinical significance.
Heteroagglutinins were a source of false-positive results when polyclonal reagents were used but are, of course, absent from monoclonal reagents.
Agglutinins Against Enzyme-Treated Red Cells
Three out of nine AHG reagents tested by Beck and co-workers (1976) reacted with enzyme-treated normal red cells, due to residual antispecies antibodies. This observation underlines the need to use suitable controls when testing enzyme-treated red cells with AHG reagents, for example when testing C3d cells produced by enzyme treatment of C3b cells. Under these circumstances, the positive reaction may be due to agglutinins for enzyme-treated red cells rather than to anti-C3d (Beck et al. 1976).
Automation of Serological Tests
The inducement to use mechanized methods of blood grouping (McNeil et al. 1963) came from the hope of introducing greater reliability and of avoiding drudgery. These hopes were largely realized with the development of machines such as the Technicon Autogrouper and the Kontron Groupamatic. The Autogrouper used a continuous flow system and comprised a dozen or more channels simultaneously, each with a different antiserum or reagent red cell. The Groupamatic used individual wells for each reaction between red cells and antibody. These machines have now largely been superseded by the Olympus PK 7200, which, like the Groupamatic, uses individual wells for each reaction between red cells and antibody. All the foregoing machines are controlled by a computer; manual intervention is not required, greatly reducing the chance of transcription errors. The machines are designed to deal with large numbers of blood samples and are not suitable for the average hospital blood bank. Most UK hospital blood banks with full automation use gel methods for both grouping and antibody screening. A minority use solid-phase microplate systems (R Knight, personal communication).
Continuous Flow Systems
In these systems, antisera and red cells are pumped continuously through coils, where agglutination is recognized and the result recorded as positive or negative. Individual samples are automatically sampled and kept separate with air bubbles. Using bar-coded samples, the computer matches the result to the corresponding sample.
Two main methods have been used, either with the large Autogroupers or with single-channel autoanalyzers: in the first the red cells are treated with a protease, e.g. 0.1% bromelin, and a polymer is added to the reaction medium, 0.25% PVP, K-9 (Rosenfield et al. 1964) or methylcellulose (Marsh et al. 1968); in the second, a low-ionic-strength medium is used, together with polybrene (Lalezari 1968). A disadvantage of the low-ionic-strength method is that it fails to detect some examples of anti-K. The autoanalyzer is no longer widely used for antibody quantification.
Systems Using Individual Reaction Wells
The Kontron Groupamatic machines are fully automated instruments that identify bar-coded samples, perform ABO and Rh grouping, detect antibodies and print out results automatically. Once enzyme-treated red cells and plasma have been dispensed into each of a series of wells on a disc, the disc is agitated to mix the contents of the wells, centrifuged and remixed. Reactions are read automatically by a simple photometric system (Matté 1971).
In the Olympus machines (PK7100 and PK7200), which are also fully automatic, the reactions take place in special microplates, each containing 120 wells with a stepped profile: agglutinates adhere to the sides of the well but unagglutinated cells fall to the bottom and form a button. The reactions are interpreted by a simple photometric system (PK7100) or by image analysis (PK7200). These systems have several advantages: <25 µl of reactants are needed. Using monoclonal reagents, tests for many red cell antigens can be made successfully, weak antigens being well detected.
Another automated system utilizing microplates employs erythrocyte-magnetized technology (EMT). In this system the patient’s red cells are pre-mixed with a magnetizing solution of iron chloride before addition to wells precoated with blood typing antibody. When exposed to a magnetic plate, the magnetized red cells form a pellet at the bottom of the well so there is no need for centrifugation or washing. After shaking to resuspend unagglutinated cells, agglutinated cells can be detected. Reverse ABO grouping and antibody screening is achieved using reagent red cells precoated with paramagnetic polymer beads (Bouix et al. 2008; Schoenfeld et al. 2009a).
Gel Systems and Semi-Automated Microplate Systems
Semi-automated systems designed to handle and read both microplates for grouping and microtubes for antibody screening are available, as is an automated system for the DiaMed-ID (Parker et al. 1995). A semi-automated PEG–IAT microplate method was found to be comparable with conventional tube and gel tests for K, Fya and Jka typing (Diedrich et al. 2001). A semi-automated fluorescence cytometric method is described by Roback and co-workers (2004). A hospital laboratory system (FREELYS Nano) for ABO/D grouping, Rh phenotyping, K typing and antibody screening using erythrocyte-magnetized technology is described by Schoenfeld et al. (2009b).
Solid-Phase Systems
In the first-described solid-phase technique, red cells were attached to the inner surface of plastic tubes to form a monolayer. The cells were incubated with antibody-containing serum, washed and then lysed to release Hb and produce a colourless background. After incubation with anti-IgG, a 0.2% suspension of IgG- or complement-coated cells was added. In a positive reaction, the coated cells were bound to the monolayer (Rosenfield et al. 1976).
Various semi-automated solid-phase assays for ABO and Rh D grouping, antibody detection and cross-matching have been described. For red cell grouping, monoclonal anti-A, -B and -D are absorbed to microplate wells; enzyme-treated cells are added and the plates centrifuged. In positive reactions, the cells adhere to the face of the well, whereas in negative reactions the cells form a button in the centre of the well. For serum grouping, the solid phase is prepared by coating the wells with rabbit anti-human red cell antibody, which then binds enzyme-treated A and B cells. The cells are lysed and the Hb washed out. Test serum is now added and anti-A or -B, if present, binds to the red cell ghosts. Binding is demonstrated by adding indicator A or B cells, centrifuging and reading. Automated reading is performed at 405 nm using a spectrophotometer (ELISA reader), with the light beam offset to pass 1.5 mm from the centre of the well in order to avoid the cell button in negative reactions. In tests on over 2000 donors there were very few discrepancies compared with conventional agglutination techniques, and these could be resolved by the use of computer-aided image analysis (Sinor et al. 1985).
In an alternative approach, the detection of haemoglobin peroxidase in adherent cells is used as an indicator system for ABO and D grouping (Moore 1984). Using this method with wells optimally coated with IgM monoclonal anti-A, anti-B or anti-D, it is possible to group cells without the need for enzyme pretreatment (Scott 1991).
The solid-phase assay with red cell adherence is also applicable to antibody screening and compatibility testing (Rachel et al. 1985). Red cells are incubated with the patient’s serum, washed and then suspended in antiglobulin serum. The suspension is transferred to IgG-coated wells, centrifuged and read visually or photometrically. A disadvantage of this test is that only IgG antibodies are detected and that there is no reaction with anti-complement to facilitate the detection of complement-binding IgG antibodies. This disadvantage can be overcome by coating the plates with AB serum in which complement has been activated with heat-aggregated immunoglobulin to ensure that the AB serum contains a sufficient amount of C3d (Guigner et al. 1988). Alternatively (as in the Biotest Solid Screen System) the plate is coated with antibodies against the anti-IgG (e.g. anti-rabbit). In a positive reaction, sensitized red cells that have been incubated with anti-IgG adhere to the solid phase. In a development of this test (Solidscreen II), an activated plate with a high affinity for the AHG reagent, detecting both IgG and C3-coated red cells, is used (Uthemann and Poschmann 1990). In comparing two solid-phase techniques, the Capture-R (CR) and the Capture-R Reading Screen (RS) it was found that not all antibodies detected in a saline-IAT were detected by these methods. On the other hand, some clinically significant antibodies, such as anti-K, anti-Fya, anti-Jka and Mca were only detected in the CR and some (anti-Jka, anti-E) only in the RS (Haslam et al. 1995).
The problem of having to centrifuge the microplates between each of the required five to six washes can be overcome by fixing the red cells or ghosts to the wells, after which ELISA washes can be used. Suitable methods include fixation of cells by using anti-human red cell antibodies, as described above, or treating plates with poly-L-lysine or glutaraldehyde so that red cells will adhere. Systems of this kind can easily be introduced into a routine blood bank. A fully automated system using microplates to determine ABO and D types and solid-phase red cell adherence assays for antibody detection and IgG crossmatching was found to have an accuracy comparable with standard manual methods (Sandler et al. 2000).
In a very thorough investigation of factors governing the results of solid-phase blood grouping tests, it was found that the amount of antibody bound to the solid phase was correlated with sensitivity. The optimal pH for binding was about pH 7 for monoclonal anti-A and -B but was pH 9.0–10.5 for monoclonal anti-D. When concentrations of monoclonal antibodies were too high, less antibody was bound. Selection of antibodies for solid-phase techniques was shown to be essential. For further details, see Scott (1991).
A solid-phase system for testing individual samples outside the laboratory has been devised (Plapp et al. 1986). Anti-A or anti-B is bound covalently to individual nylon squares that are then attached to plastic strips to form ‘dipsticks’. A drop of blood is added to the dipstick; after 1 min the stick is rinsed with saline; a positive result is indicated by the red colour of adhering cells. From a technical point of view, this method seems likely to be more reliable than determining ABO groups on cards on which anti-A and anti-B have previously been dried (Eldon 1955). Nevertheless, objections to both systems are the same, namely that it is difficult to devise controls that are as reliable as those available when liquid blood group reagents are used, and that serum cannot be tested to confirm ABO groups.
Quantification of Red Cell Antibodies
Antibody Titres
The classical method of determining antibody ‘strength’ is to determine the titre of a serum, expressed as the reciprocal of the highest dilution of the serum that will produce a detectable reaction with selected red cells. There are several reasons why this method gives only a very approximate indication of antibody concentration. First, the method is unsound in principle as it estimates only the amount of antibody bound to red cells, not the amount of antibody in the serum. At the endpoint of the titration, agglutination is caused by the relatively small number of antibody molecules in the serum with the highest affinity, and therefore the proportion of such molecules in the serum has a considerable influence on its titre. As an example, if two sera each contain the same concentration of an antibody but one example has a binding constant 10 times higher than the other then the antibody with the higher binding constant will have a titre approximately 10 times greater than the other (Hughes-Jones 1967). Second, as usually carried out, dilution of the serum is performed in a series of double-dilution steps. This method is inaccurate and, unless special precautions are taken, traces of serum are carried over from one dilution to another. In practice, the titres of anti-D sera determined by a manual method have been found to be poorly correlated with the antibody concentrations of the same sera determined by an isotope method (Hughes-Jones 1967).
A score is a better estimate of antibody concentration than a titre. To determine a score all positive reactions obtained with serial, doubling dilutions of an antiserum are given a value based on the strength of the reaction, e.g. + + + + = 10, + + + = 8, etc. The sum of these values is the score (see Marsh 1972).
For routine determinations of antibody concentration, the autoanalyzer gives far more reliable results than those given by titration.
Use of the Autoanalyzer
A single-channel autoanalyzer, using the bromelin–methylcellulose method, provides a simple method of quantifying anti-D with fair reproducibility, for example with a coefficient of variation of 17%. Reproducibility is substantially better than that of manual titration (Judd and Jenkins 1970). Nevertheless, interlaboratory reproducibility is not very good, having a coefficient of variation of about 20% (Fleetwood and McNeill 1990).
In a comparison with the 125I antiglobulin method, two-thirds of estimates of anti-D made in the autoanalyzer agreed to within 24% (Moore and Hughes-Jones 1970).
Despite the considerable inherent assay variability the autoanalyzer was still widely used for the quantification of anti-D (Bowell et al. 1982) and anti-c (Kozlowski et al. 1995) in pregnancy and for the quantification of anti-D immunoglobulin preparations until recently.
125I-Labelled Antiglobulin
Direct Labelling of Antibody with Radio-Iodine
When whole serum is labelled with radio-iodine, lipids are labelled in addition to serum proteins. When the labelled serum is incubated with the red cells the lipids exchange rapidly between the serum and cells and mask the specific uptake of antibody (Hughes-Jones and Gardner 1962). Methods exist for greatly reducing non-specific uptake but are too tedious for routine use. Purified IgG is now used for labelling.
Use of Radio-Iodine-Labelled Anti-IgG
In this technique, 125I-labelled anti-IgG is used to estimate the number of IgG molecules on washed sensitized red cells. An advantage is that a given batch of labelled anti-IgG can be used to estimate the amount of antibody in many different sera. The technique has been used particularly in estimating the amount of anti-D in preparations intended for immunoprophylaxis, although concentrations of anti-D as low as 1 µg of anti-D/ml can be measured (Hughes-Jones and Stevenson 1968).
An assay based on the consumption of anti-IgG by sensitized red cells, the remaining anti-IgG being measured by precipitation with 125I-labelled IgG in the presence of PEG, has been described (van de Winkel et al. 1988). The reproducibility of this technique was found to be excellent.
Enzyme-Linked Immunosorbent Assay
The ELISA technique was adapted by Leikola and Perkins (1980b) for the quantification of IgG antibody on red cells. Anti-IgG was conjugated to alkaline phosphatase using glutaraldehyde and was calibrated by adding doubling dilutions to tubes containing fixed quantities of IgG; p-nitrophenyl phosphate was added as substrate; the reaction was stopped after 30 min with NaOH and the yellow colour measured at 405 nm. Similarly, the anti-IgG conjugate was added to antibody-coated red cells followed by substrate and the colour measured. It was found that the test was two to three doubling dilution steps more sensitive than a conventional antiglobulin test and that as little as 5 ng/ml of anti-D could be detected.
In a further paper, the authors described the use of the enzyme-linked antiglobulin test (ELAT) to study the uptake of antibodies on to red cells suspended in LISS (Leikola and Perkins 1980a). ELAT has been used to detect fetal D-positive cells in the circulation of D-negative women; the lowest concentration of D-positive cells that could be detected reliably corresponded to a transplacental haemorrhage of about 6 ml of red cells (Riley et al. 1982).
ELATs have also been used to quantify IgG, IgM, IgA or C3 on the red cells of patients with AIHA (Kiruba and Han 1988; Sokol et al. 1988).
A method for quantification of anti-D in alloimmunized patients and in anti-D immunoglobulin preparations using ELISA is described (Lambin et al. 1998; Ahaded et al. 1999). Red cells (10% suspension) are sensitized with anti-D (<1.2 µg/ml) at 37°C for 2 h, the antibody-D polypeptide immune complexes solubilized in the non-ionic detergent Triton X-100 (20–24°C for 30 min) and recovered from the supernatant after centrifugation. The immune complexes are then captured in microplates coated with rabbit anti-mouse IgG Fc and monoclonal mouse anti-human IgG Fc. Binding of human anti-D is measured using alkaline phosphatase-labelled rabbit anti-human IgG. The method can be modified for quantification of IgG subclasses by substitution of mouse anti-human IgG subclass-specific monoclonal antibodies for the mouse anti-human IgG Fc. These authors report an excellent correlation between their ELISA and autoanalyser measurements and consider the method to be more sensitive than flow cytometric methods.
A competitive enzyme-linked immunoassay (EIA) for the quantification of monoclonal anti-D for prophylaxis has also been described (Thorpe et al. 2003a). Fifty monoclonal anti-D were quantified by inhibition of red cell binding of biotinylated monoclonal anti-D (BRAD-5) and their potencies estimated against the international reference preparation (IRP) for anti-D Ig. Two antibodies were insufficiently inhibitory and so could not be quantified and 11 showed dose responses not parallel to those of the other monoclonal antibodies and the IRP. These authors conclude that competitive EIA is suitable for quantifying most monoclonal anti-D.
Flow Cytometry
A flow cytometer provides delivery of a single file of cells or other particles in suspension through a focused laser beam. Signals due to light scattering and/or emission of fluorescence are available for analysis or may be used as a basis for cell sorting.
Flow cytometry has been used to measure the amount of antibody bound to red cells (DeBruin et al. 1983), to estimate the amount of antibody of different samples of coated red cells giving maximum agglutination in the antiglobulin test (Nance and Garratty 1984), to estimate the density of antigen sites on red cells (Langlois et al. 1985; Bockstaele et al. 1986; McHugh et al. 1987), to analyse the distribution of c, D and E epitopes; to measure the relative density of D sites on selected samples of red cells and to quantify the relative numbers of cell-bound IgG1 and IgG3 anti-D molecules (see Chapter 5); to study the relative numbers of IgG1 and IgG3 anti-D molecules in anti-D preparations used for immunoprophylaxis and to assess the degree of red cell sensitization by IgG1 and IgG3 anti-D monoclonals in experimental studies (see Chapter 5) and in studies relating IgG1 and IgG3 anti-D concentrations to the severity of HDN (see Chapter 12). Flow cytometry has also been used to measure the survival of compatible and incompatible red cells (see Chapters 9 and 10) and to detect fetal D-positive red cells in the circulation of D-negative women (see Chapter 12).
Austin and McIntosh (2000) compared five methods for quantification of 10 serum samples containing anti-D by flow cytometry. Plotting the mean anti-D value for each sample as a percentage of the value determined by autoanalyzer revealed wide variability between the methods. These authors concluded that further validation of the flow cytometric method was required before routine application. In a subsequent study, a flow cytometric assay was compared with autoanalyzer quantification with more favourable results (flow cytometry gave values ranging from 87% to 129% of the value obtained by autoanalyzer). It was concluded that flow cytometry is a suitable method for quantifying polyclonal anti-D immunoglobulins (Schaffner et al. 2003).
Relative Sensitivity of Different Methods of Antibody Detection
The Indirect Antiglobulin Test
The lowest concentration of IgG anti-D detectable by the standard IAT is about 10 ng/ml; using this test in external quality assessment surveys 98% of unselected participants were able to detect 11 ng/ml (Pinkerton et al. 1984).
ELAT was found to be distinctly more sensitive than the IAT, the lowest concentrations detectable being 4 and 10 ng/ml respectively (Postoway et al. 1985). In one recent survey, 100% of participants detected anti-D at a concentration of 0.1 IU/ml, i.e. 0.02 µg/ml (UK NEQUAS for blood group serology).
Other Manual Methods
Two-stage tests with enzyme-treated cells are generally believed to be more sensitive than the IAT in detecting IgG anti-D, although no evidence in support of this view was obtained in proficiency testing described by Pinkerton and co-workers (1984).
The manual low-ionic-strength polybrene test is capable of detecting about 1 ng of anti-D/ml without an antiglobulin stage (M Contreras, unpublished observations). Although no quantitative studies have been done, it seems that the PEG antiglobulin test is even more sensitive. The titre of 10 out of 11 alloantibodies was found to be higher in the PEG test than in the polybrene test (Nance and Garratty 1987).
The Autoanalyzer
Using the low-ionic-strength polybrene method, or the bromelin–methyl cellulose method, the lowest concentration of IgG anti-D detectable is approximately 2 ng/ml.
Reference Preparations
Lyophilized monoclonal IgM anti-A, anti-B and anti-D preparations are available as reference preparations from the World Health Organization and can be used for determining minimal acceptable levels of potency of reagents (Thorpe et al. 2006a,b). The first anti-D immunoglobulin reference preparation (68/419) contained 60 µg of anti-D; 1 µg is equivalent to 5 IU (Gunson et al. 1980). Comparative studies of this preparation, a national standard, two commercial and two national preparations for clinical use showed that the isotope method, manual haemagglutination and the autoanalyzer all gave similar values (Bangham et al. 1978). A lyophilized anti-D immunoglobulin preparation (01/572) was adopted by WHO as the second international standard for anti-D immunoglobulin with an assigned potency of 285 IU/ampoule (Thorpe et al. 2003b). The new standard was calibrated against 68/419 and two other preparations in a collaborative international study involving 25 laboratories in 15 countries. Most laboratories (20 out of 25) assayed the preparations using an autoanalyzer, competitive enzyme-linked immunoassay (see above) or flow cytometry. The new standard was designated the standard for anti-D immunoglobulin, Lot 4 by the United States Food and Drug Administration Center for Biologics Evaluation and Research and as the first Biological Reference Preparation for anti-D immunoglobulin by the European Directorate for the Quality of Medicines. In view of limited supplies of reference preparations, individual countries and laboratories should set up their own working standards after determining the relation of such standards to the international reference preparation. For up-to-date information about the availability of WHO standards see http://www.nibsc.ac.uk/biological.html.
A lyophilized anti-D reference preparation for standardizing enzyme activity has been developed by the ISBT/ICSH.
Polyspecific reference antiglobulin reagents available from the ISBT and ICSH have been mentioned above.
Red cells with defined levels of expression of A and B antigens have been prepared by incubating group O cells with synthetic A or B-active glycolipids for use in the quality control of blood grouping (Henry 2009).
The Selection of Compatible Red Cells
The Serological Crossmatch
In selecting compatible blood, the traditional practice is first to determine the patient’s ABO and Rh D groups, second to ‘screen’ the patient’s serum for red cell antibodies other than anti-A and anti-B and, third, having selected blood of the same ABO and Rh D group, to crossmatch the donor’s red cells against the recipient’s serum. These three steps will be considered in turn.
Determination of the Recipient’s ABO and RhD Groups
The determination of the ABO groups differs from the determination of other blood groups in two ways. First, the ABO system is by far the most important blood group system in transfusion practice; second, the ABO system is the only one in which the antigens present on the red cells can usually be predicted from a knowledge of the antibodies present in the serum.
The standard method of determining the ABO group of a sample is therefore to test the red cells against monoclonal anti-A and anti-B and to test the serum against group A and group B red cells. Traditional practice includes testing the red cells against anti-A,B to detect weak group A samples that are not agglutinated by anti-A from a group B donor but this is not necessary when monoclonal anti-A selected to react with weak A is used. Monoclonal anti-B used for ABO grouping should not react with acquired B antigen. In grouping recipients, it is not necessary to recognize weak groups of A; recipients with weak group A may be safely transfused with group O blood.
It is usual to test the serum against A1 rather than A2 cells, as A1 cells have more A antigen and thus serve better to detect anti-A. In some subjects of subgroups A2 and A2B, the serum contains anti-A1; in that case, the serum has to be retested to demonstrate that it will not react with A2 cells. Testing for ABO antibodies in the serum can be omitted for neonates or if a secure automation system is used. In the latter case, there must be at least one historic record where the results of testing for serum antibodies is recorded and the current cell group must be identical with all historical records (Chapman et al. 2004).
When speed is essential and when a tube test is used, the mixtures can be centrifuged immediately. For rapid RhD grouping, IgM monoclonal anti-D reagents are available for tube or slide tests. When tests have to be done in a hurry it is more convenient to use blood mixed with an anticoagulant than clotted blood because with the clotted samples there may be delay in separating the clot and obtaining the red cells from the bottom of the tube.
In the absence of a secure automation system two IgM monoclonal anti-D reagents not reactive with DVI cells (see Chapter 5) should be used to determine the D phenotype (Chapman et al. 2004). Most monoclonals fail to react with one or more of the D variants so it is advisable to use a combination of two monoclonals, which together recognize all common variants in the population being tested.
In determining the patient’s D group, the risk of failing to detect weak or partial D should be ignored. If a recipient with a weak D antigen is misclassified as D-negative and transfused with D-negative blood, no harm will be done. The IAT should not be used to detect weak D samples, as there is a substantial chance of a false-positive result (see later). Such an error might lead to the subject being transfused with D-positive blood, or, if pregnant or recently delivered, not receiving anti-D immunoglobulin.
In view of the known frequency of errors in ABO and D grouping, due mainly to clerical mistakes, it is important to minimize the risk of error by separating the procedure into distinct tasks and using different members of staff to perform each task (e.g. separating the documentation of reaction patterns from the final interpretation; Chapman et al. 2004).
Whenever a patient’s blood groups are determined, the results should be recorded so that if further samples from the same patient are tested the results can be compared.
Positive and negative controls should be used on a regular basis.
Error Rates in Blood Grouping
Proficiency testing schemes have shown how difficult it is to eliminate errors in ABO and RhD grouping. Results on samples sent to some 2000 laboratories indicated that the average error rate was 0.64% (Myhre et al. 1977). In trials carried out in Canada over a 4-year period, the frequency of major errors in ABO grouping was between 0.13% and 0.14%; errors in RhD typing fell from 0.66% to 0.23% over the period (Pinkerton et al. 1979, 1981).
In a review of the UK National External Quality Assessment Scheme over the period 1985–2000 it was found that error rates for ABO grouping fell from 0.19% in 1984/1985 to 0.05% in 1998/2000. This improvement was attributed to the elimination of technical errors in misgrouping A2B samples as group B, with the residual errors being due to transcription, transposition or misinterpretation. In contrast, error rates for RhD grouping did not change significantly over the same period (0.25% in 1984/85 and 0.21% in 1998/2000), at least 70% of the errors in 1998/2000 were the result of transcription or transposition (Knowles et al. 2002). These findings emphasize the importance of involving two members of staff in the overall process of documentation and interpretation of grouping results.
In an attempt to reduce errors leading to incompatible transfusions, some institutions have adopted a policy requiring verification of patients ABO and Rh type on two separate blood samples (reviewed by Mintz and Dzik 2009). Figueroa et al. (2006) describe a policy which requires a second sample to be tested for all previously untyped non-group O patients likely to require transfusion to avoid miscollected wrong blood in tube (WBIT) samples. They detected 94 WBIT errors over a 17-year period and averted five potential ABO-incompatible transfusions. In the event that two blood samples have not been tested, group O blood is provided for transfusion. Goodnough et al. (2009) report on introduction of a similar policy whereby the patients ABO and Rh type must be verified on two separate blood samples before transfusion. The value of having a single database of patient transfusion data for local areas encompassing several hospitals is illustrated by Macivor et al. (2009). These authors compared historical ABO typing results for individual patients in the records of 16 hospitals with current typing results over a period of 30 months and identified six discrepancies which in three cases (correct type O, current type A), would have resulted in ABO incompatible transfusions.
Screening Tests on the Serum of Potential Recipients
The advantages of screening the serum of potential recipients for the presence of alloantibodies are as follows. First, when the screening test can be carried out well in advance of transfusion and when alloantibodies are detected there will be time to identify the antibody and select compatible units. Second, special red cells can be used for the screening test, which have been obtained from donors homozygous for alleles such as c, E, Jka and Fya; such red cells react far more strongly than cells from heterozygotes and their use is essential for the detection of some weak alloantibodies. It is virtually impossible to find a single donor who is homozygous for all the relevant alleles and the usual practice is to select two to three donors who between them are homozygous for the most important alleles such as c and Jka. In screening sera for alloantibodies, different samples of red cells should not be pooled, as this may lead to failure to detect antibodies (De Silva and Contreras 1985; Knowles et al. 2002).
In screening, the LISS antiglobulin test is considered the most suitable method (Knowles et al. 2002; Chapman et al. 2004). The PEG–IAT and the IAT in microplates are also considered to be as sensitive. However + or ++ reactions may easily be missed in spin-tube IATs due to inappropriate reading, particularly in large-scale screening (see Engelfriet and Reesink 1995). Great advantages of gel/column agglutination techniques are that reading is much easier and that results can be checked by others.
For pregnant women and patients who have been transfused recently, who could be developing an immune response, a second test, such as the MPT, should be used in addition.
In patients who are to undergo operations involving induced hypothermia there is no need to screen the serum at a temperature below 37°C. No convincing account has ever been published of red cell destruction caused, during hypothermia, by a red cell alloantibody active in vitro only at a temperature below 37°C.
Antibodies Passively Transferred by Injections of Immunoglobulin
D-negative patients receiving injections of anti-D immunoglobulin may, of course, have detectable amounts of anti-D in their plasma for considerable periods after injection. They may also have detectable amounts of antibodies of other specificities if these are present in sufficient concentration in the immunoglobulin. In one such case, passively transferred anti-E, anti-Leb and anti-HI, all IgG, were detected in a patient who had received 900 µg of anti-D immunoglobulin (Wright et al. 1979).
Errors in Antibody Detection
Two kinds of error may be distinguished: those due to technical errors and those inherent in the techniques used. As an example of the first kind, one may take the results of quality control exercises in which participants used various techniques.
In a comparative study carried out in Canada and the UK about 17% of participants failed to detect anti-D at 0.1 IU (0.02 µg) per millilitre and about 1.5% failed to detect anti-D at 0.25 IU (0.05 µg) per ml (Pinkerton et al. 1985). In the UK between 1991 and 1998 a progressive improvement in the performance of the IAT was noted such that by 1998 100% of participants detected anti-D at a concentration of 0.1 IU (Knowles et al. 2002).
In the UK, error rates for false-negative antibody screens have fallen from 3.2% in 1984/1985 to 0.5% in 1998/2000. In contrast with ABO and D typing the majority of these errors result from technical problems relating to test sensitivity and have been lower with the antiglobulin method than with other methods. Thus, in detecting anti-D at a concentration of 0.05 µg/ml error rates were as follows: antiglobulin test, 2–3%; with an albumin method, 14–21%; and using enzyme-treated cells 27–49% (Holburn and Prior 1986). Although most laboratories used a one-stage enzyme technique, even two-stage enzyme techniques proved to be less reliable than antiglobulin techniques. Nevertheless, the incidence of false-positive results was significantly higher in the antiglobulin test than in any other technique (Holburn and Prior 1984, 1986). By 1999 most laboratories in the UK had abandoned the use of enzyme-treated cells and direct saline agglutination tests relying solely on the indirect antiglobulin method. In the same period, traditional tube technology was largely replaced by the use of gel/column methods (see Figure 8.1). When cells heterozygous for Jka are used for antibody screening using gel/column technology anti-Jka may be missed (Knowles et al. 2002).
Selection of Donor Blood (Red Cells)
In countries where blood is collected at transfusion centres, most hospitals rely entirely on the description on the label of the unit of blood and make no further tests on it. In the UK, for example, it is not considered necessary to verify the ABO and D groups of units received from a Blood Transfusion Centre. In the USA, the ABO group of all units and the D group of all D-negative units must be verified.
Limitation of the supply of D-negative blood very occasionally makes it necessary to consider giving D-positive blood to D-negative recipients. If the recipient is a male who has not been transfused before, and particularly if the recipient is elderly, the transfusion of D-positive blood is not likely to lead to trouble. Anti-D is not likely to be formed for several weeks after the transfusion, so that if further transfusions are needed within a few days of the first transfusion more D-positive blood can be given (provided that crossmatching is carried out with a sample obtained on the day of transfusion). Although it may be defensible to give D-positive blood to D-negative males, it must be an absolute rule that D-positive blood is never transfused to a D-negative female who is still capable of childbearing.
In a D-negative woman who has reached the menopause, the transfusion of D-positive blood is much less safe than in a D-negative man because the woman may have been sensitized by pregnancy. Such a case, in which a woman developed a severe DHTR after her first transfusion, is described in Chapter 11. Donations for D-negative patients immunized to D must be C negative and E negative as well as D negative, because subjects who make anti-D often make anti-C(G) or anti-E as well. However, for D-negative patients who have not made anti-D it is sufficient to give blood that is D negative and the donor red cells need not also be C negative and E negative (compare with Chapter 5).
When alloantibodies active at 37°C, other than anti-A, -B and -D are present, or have been present in the past but are no longer detectable, for example anti-Jka, red cells lacking the corresponding antigens must be selected for transfusion.
Is It Practicable to Try to Prevent Immunization to Antigens Other Than D?
In Females Who Have Not Yet Reached the Menopause
It seems worth giving serious consideration to the possibility of trying to prevent the formation of anti-K and anti-c. It is relatively easy to avoid using K-positive blood for K-negative recipients, but finding c-negative blood for c-negative recipients involves a substantial extra amount of work. Nevertheless, as haemolytic disease of the newborn due to anti-D becomes less common, the possibility of preventing immunization of women by c may seem worth pursuing. High throughput automated DNA-based methods of blood typing to screen large donor databases provide a method for achieving such a goal (Denomme and van Oene 2005; Hashmi et al. 2005, 2007; Avent et al. 2007).
In Patients Who Receive Multiple Transfusions
It is very likely that antibodies will be made to the more immunogenic of the red cell alloantigens that the patients themselves lack. For patients with sickle cell disease or thalassaemia, a strong case has been made for using red cells matched for Rh antigens (C, c, E, e) and K in addition to ABO and D. A retrospective analysis of 137 alloimmunized patients with sickle cell disease in the USA demonstrated that production of all alloantibodies would have been prevented for more than one-half of these patients if matching for these antigens had been undertaken (Castro et al. 2002). As approximately 13.6% of random white blood donors would be expected to match for these antigens, these authors concluded that such an approach is feasible. They also noted that extending the matching further to include S, Fya and Jkb would have prevented alloimmunization in 70% of patients, but as only 0.6% of white donors in the USA would match such a protocol this approach did not represent a realistic strategy. In a different study, where extended antigen matching was not performed it was concluded that delayed serological and/or haemolytic transfusion reactions occurring in alloimmunized sickle cell patients (29% of paediatric patients and 47% of adults) did not result in severe clinical outcome ‘in most instances’ (Aygun et al. 2002). A survey of 37 academic medical centres in the USA and Canada revealed that 73% routinely provide antigen-matched blood for sickle cell patients. Of these, 89% match for C, E and K (Afenyi-Annan et al. 2004). Much of the difficulty in providing extensive antigen-matching blood for patients with sickle cell disease in Europe and the USA stems from the relatively small number of black donors in these countries. A comparison of the frequency of alloantibodies in 190 sickle cell patients in Jamaica compared with 37 patients in the UK revealed a striking difference. Only 2.6% of Jamaicans transfused had alloantibodies in comparison with 76% of those transfused in the UK. Multiple anti-bodies occurred in 63% of UK residents but in no Jamaicans (Olujohungbe et al. 2001). The potential utility of high throughput automated DNA-based methods of blood typing to ensure these patients get the most compatible blood available from the donor inventory has generated great interest, but it must be recognized that the efficacy of such an approach is severely limited when there is an imbalance between the ethnic groups represented in the donor and patient populations (Anstee 2009).
Screening of the Donor’s Plasma
When group O whole blood is transfused to recipients of groups A, B or AB and when the donor’s plasma contains potent anti-A,B, a haemolytic reaction may be produced (see Chapters 10 and 11). The risk is diminished when plasma-reduced blood is used and further diminished by the use of red cell concentrates or red cells suspended in additive solutions.
In some hospitals it is the custom to give group O blood (O, D-negative blood for female recipients or for previously transfused males) in grave emergencies without any compatibility tests. When this is done, the risk of producing a haemolytic reaction from anti-A or anti-B in the donor’s plasma can be greatly reduced by selecting group O donors with low-titre antibodies. A commonly used test is to screen for high-titre anti-A,B using A1B cells suspended in saline containing AB substance. Donors whose serum gives a positive reaction in this test are considered to be dangerous if their blood is transfused to patients of groups other than O. This test can be performed in parallel with automated ABO and D grouping. Many transfusion services put a special label on group O units with high-titre anti-A or anti-B, to indicate that these units should be used only for group O recipients.
There is some evidence that a test for anti-A and anti-B lysins might be better than a test for high-titre agglutinins in excluding ‘dangerous’ group O donors (Grove-Rasmussen et al. 1953; fifth edition, p. 530).
Very few haemolytic reactions due to the presence in the donor’s plasma of antibodies other than anti-A and anti-B have been described (see Chapter 11). The increasing use of red cells suspended in additive solutions is likely to make such accidents even rarer in the future. In screening the plasma of donors, a relatively simple test, capable of detecting potent antibodies to the most clinically important antigens, is needed. In large donor centres, screening is undertaken on automated machines. Pools of six donor sera can be safely used in the solid-phase Immunocor-Capture ™-R system (Beck et al. 1991).
The results of screening the serum of more than 84 000 blood donors during 1975 were described by Giblett (1977). The overall frequency of antibodies was relatively low, i.e. 0.32%, due partly to the fact that the average age of the donors was only about 30 years. Of the antibodies, 68% were anti-D. In the donors with no history of transfusion or pregnancy the frequency of antibodies was 0.04%. It was pointed out that by testing the serum of donors giving a history of previous transfusion or pregnancy, and by also testing the serum of all D-negative donors, only about 1 in 10 000 sensitized donors would be undetected. In view of the fact that the undetected antibodies would virtually always be of low titre it seems quite unjustified to devote much effort to their detection. Certainly, the use of the antiglobulin test in these circumstances seems very wasteful of time and money (Giblett 1977).
Crossmatching Tests
Ottenberg (1908) was the first to apply Landsteiner’s discovery of the blood groups to transfusion practice by testing for lysis and agglutination of donor’s red cells by the recipient’s serum as a preliminary to transfusion. During the following few years it was found that in over 100 cases in which the patient’s serum did not lyse the donor’s red cells no haemoglobinuria developed. By contrast, when there was lysis in vitro in the ‘cross’ test (as it was then called – see Ottenberg 1937), there was always some intravascular lysis in the recipient (Ottenberg and Kaliski 1913). Crossmatching tests capable of detecting incomplete antibodies became widely used only in the mid-1940s, after the discovery of the effects of suspending red cells in albumin and of using antiglobulin serum. However, a test in which whole citrated blood from donor and recipient was mixed in various proportions was introduced much earlier by Weil (1915). As pointed out by Rosenfield (1977), the use of this test may have first revealed RhD antibodies in human sera, as the test was positive in five cases of serious reactions following ABO compatible transfusions described by Unger (1921).
As described above, it is standard practice to screen the serum of potential recipients for red cell alloantibodies. When this has been done, an abbreviated crossmatch procedure can be used, of which the main purpose is to detect ABO incompatibility due, for example, to misidentification of the patient (see below). When preliminary screening has not been done, the test used for crossmatching must be capable of detecting virtually all clinically significant antibodies. In the detection of red cell alloantibodies the IAT is undoubtedly the best single test available. As discussed earlier, the ratio of serum to cells is very important in determining sensitivity; if possible, not less than four volumes of serum should be mixed with one volume of a 2–5% suspension of red cells in isotonic saline. After 3 min at room temperature, the tube is centrifuged; the supernatant is examined for lysis and the cells are then resuspended and examined for agglutination. If the test is negative at this stage the tube is incubated at 37°C for at least 30 min; the cells are then washed four times, antiglobulin serum is added, the tube centrifuged once more and the cells examined for agglutination. When the test is carried out at low ionic strength the incubation time can be reduced to 10 min (for further details, see earlier section on the anti-globulin test).
In patients who are to undergo surgery involving hypothermia, crossmatching tests (like screening tests) should be carried out, as usual, at 37°C.
Type and Screen
In many blood transfusion laboratories a great many units of blood (or red cell concentrates) are crossmatched each day for patients who are most unlikely to require transfusion, for example patients who are to undergo a surgical procedure in which it is unusual for blood to be transfused. To avoid units of blood being reserved unnecessarily the policy was adopted in such cases of simply grouping the patient’s red cells and screening the serum for abnormal antibodies, i.e. type and screen (Henry et al. 1977). The fact that the patient’s serum has been screened for unexpected antibodies should ensure almost complete transfusion safety (Boral and Henry 1977). To exclude ABO incompatibility either an abbreviated crossmatch is done or the ABO groups of donors and patient are checked before transfusion (see below). The type and screen procedure is now not only used for patients who are to undergo an elective surgical procedure but, in many hospitals, for all patients except (1) patients in whose blood abnormal red cell antibodies are or have been detectable; (2) patients who have received multiple blood transfusions; and (3) newborn infants. For these patients a full crossmatch including an indirect antiglobulin test should always be carried out.
The concept of a maximum surgical blood order schedule (MSBOS) has proved extremely valuable. MSBOS is a list of elective surgical procedures, together with the number of blood units required, based on a retrospective analysis of actual usage in each hospital. A satisfactory order meets the requirements of 90% of patients (Friedman 1979). For a detailed discussion, see Standard Haematology Practice (1991).
Abbreviated Crossmatching (Immediate-Spin Vs. Computer Issue)
As mentioned above, when typing and screening is not followed by a crossmatch including an IAT, in order to exclude ABO incompatibility, either an abbreviated crossmatch must be done or the donor’s and patient’s ABO groups must be checked with anti-A and anti-B reagents.
An abbreviated crossmatch can comprise the so-called immediate-spin or non-serological methods such as electronic issue. The UK BCSH recommend computer issue without a serological crossmatch as the method of choice when a laboratory wishes to replace the IAT crossmatch with an alternative method. This is because of concerns about lack of standardization of the immediate-spin method (O’Hagan et al. 1999).
The immediate-spin method is simply to mix a saline suspension of donor red cells with recipient’s serum in a tube, centrifuge immediately and examine for agglutination. The justification for using only an abbreviated crossmatch when the patient’s serum has already been screened for antibodies lies in the observation that very few antibodies are detected by rigorous crossmatching that have not been detected by screening. For example, Heistø (1979) reported the results of screening more than 20 000 patients by rather an elaborate technique, involving an IAT with two different cells containing most of the red cell alloantigens and a papain technique against R1R2 cells, which revealed alloantibodies in approximately 0.75% of the patients. Only 5030 of the patients became recipients and, in these, compatibility tests revealed previously undetected antibodies in only three cases (0.06%); all three antibodies were very weak. Results reported by Oberman and co-workers (1978) were similar and led the authors to conclude that if blood that had been carefully screened by the antiglobulin test was subsequently released, after a crossmatch consisting only of an immediate spin, there would have been only about a 0.06% chance that an extremely weak antibody of potential clinical significance would have gone undetected.
The conclusions of Walker (1982) were almost identical. He calculated that the risk that an antibody to a low-frequency antigen, missed on screening, might be the cause of incompatibility because it was missed on subsequent abbreviated crossmatching was approximately 0.06% and that such an event would very seldom be of clinical significance because most of the antibodies concerned cause little or no red cell destruction. Although these calculations led Walker (1982) to conclude that there was no need to do an antiglobulin crossmatching test for patients about to undergo surgery who were unlikely to require a blood transfusion, he stated that he would like to retain the antiglobulin test in routine crossmatching procedures for patients who actually needed blood.
In a later study, some 47 000 crossmatches were performed for about 17 500 patients, whose serum was also screened for alloantibodies. Of 284 clinically significant antibodies, 30 were detected only during crossmatching, using the IAT. Of the 30 antibodies, 24 were directed to low-frequency antigens but were otherwise unidentified; the remaining six had the following specificities: anti-CW (two cases), -Kpa, -M, -Mia and -Jka. In view of the relatively high incidence of antibodies to low-frequency antigens detected only by crossmatching, together with the relatively low cost of retaining the antiglobulin test, the authors concluded that the antiglobulin test should be retained for crossmatching (Motschman et al. 1985).
To replace the immediate-spin crossmatch a computer crossmatch can be used. The computer crossmatch is performed only for patients without unexpected antibodies in the past or present and with a record of two (or more) concordant results of ABO and Rh D typing (Butch et al. 1994). Furthermore, it requires software that does not permit the issue of ABO-incompatible blood (Judd 1998; Chapman et al. 2000). Computer crossmatching can result in a considerable reduction in blood wastage and significant savings in laboratory workload (Cox et al. 1997; Miyata et al. 2004).
Electronic Remote Blood Issue
The speed with which blood can be provided to patients eligible to receive blood via computer crossmatch can be greatly increased by electronic remote blood issue (ERBI). ERBI allows the issue of blood from a blood refrigerator remote from the blood bank. Staves et al. (2008) found that blood could be made available to cardiac surgery patients within 30 sec to 2 min using this method. and that unused requests for blood were reduced from 42 to 20%.
Errors in Abbreviated Crossmatching
Problems in detecting ABO incompatibility by the immediate spin have been reported. When an immediate spin was used, or a spin after a very short period of incubation, the reaction between A2B cells and B serum was missed in about 40% of cases although other ABO-incompatible combinations were detected reliably (Berry-Dortch et al. 1985).
Using an immediate spin in 1000 combinations of patients with blood group B and donors with blood group A2B, incompatibility was detected in only 40% of the cases using a test in saline and in 64.4% using LISS. Incompatibility was also missed sometimes with other combinations (Shulman et al. 1987). These findings do not seem to be very worrying because mis-grouping of A2B donor units as B is rare.
When the centrifugation step in the immediate-spin crossmatch was delayed for 2 min, no agglutination and only weak to moderate lysis was seen in 5 out of 200 crossmatches between group O serum and group A1 red cells. The absence of agglutination was presumed to be due to steric hindrance by C1 fixed on the red cells. In all five cases, agglutination occurred when the red cells were suspended in saline containing EDTA (Shulman and Calderon 1991). Failure to detect ABO incompatibility when centrifugation is not delayed may be due to a prozone effect if the antibodies in the patient’s serum are exceptionally strong, probably because C1 is immediately fixed on the red cells (Judd et al. 1988).
Using a polybrene method, incompatibility between A2 and anti-A, and B and anti-B, was missed in a substantial number of cases (Mintz and Anderson 1985), presumably because IgM antibodies are less reactive in polybrene. In contrast to these findings, when red cells suspended in LISS were incubated with serum at 37°C for 10 min before being spun, only one example of anti-A (and none of anti-B) was missed in the course of testing nearly 3000 sera (Trudeau et al. 1981).
It seems that the most reliable method of all, in looking for ABO incompatibility, is to follow a test for agglutination by the antiglobulin test; the increased sensitivity may be due partly to the longer period for which the red cells are incubated and partly to the potential for detecting bound complement.
Another reason for retaining the antiglobulin test in crossmatching is simply that because in practice there are so many potential sources of error, some redundancy seems desirable. At the same time, it is clear that if the IAT is omitted from crossmatching, in patients in whom no antibodies have been found on screening, the risk of giving incompatible blood will be extremely small.
Crossmatching in Patients Having Repeated Transfusions
In patients who are having blood transfusions every day or two there is a temptation to obtain a large sample of serum and use this for crossmatching for several successive transfusions. One must remember that any one of a series of transfusions may stimulate a secondary response in a previously immunized subject and that the antibody may rapidly increase in titre once it has appeared. In order to minimize the risk of failing to detect newly formed alloantibodies, one should insist on having serum taken not more than 24 h before the next proposed transfusion.
On the other hand, if incompatible red cells are transfused to a patient whose serum contains a weak antibody, all the antibody may be temporarily absorbed so that if the patient requires a further transfusion, serum taken before the first transfusion may be best for crossmatching. Most cases of this kind can be diagnosed by carrying out a DAT on the patient’s red cells at the time of the second or later transfusions provided that enough sensitized donor cells are still present.
If a previously identified antibody has been temporarily absorbed or is no longer detectable, donors who are negative for the corresponding antigen must be selected. For example, if a patient is known to have had anti-c but the antibody is no longer detectable, the patient should receive blood from donors known to be c negative.
A suggested routine for patients receiving a series of transfusions is to screen the pre-transfusion serum and all subsequent serum samples against red cells containing the common red cell antigens (see above). The pre-transfusion samples of red cells should be taken into a suitable preservative solution and stored at 4°C in case further blood grouping tests are needed; in identifying any antibodies that the patient may form, it is extremely helpful to know the patient’s red cell phenotype and it is evidently far easier to determine this on a pre-transfusion sample than on one taken after the patient has received many units of blood. Molecular methods may be useful for blood typing the patient post transfusion (see ‘Molecular methods of red cell grouping’).
Compatibility Tests in Newborn Infants
At birth the only alloantibodies present in the sera of most infants are those that have been transferred across the placenta from the mother’s circulation. In the occasional cases in which alloagglutinins formed by the infant are present, they are found in only trace amounts. It can be assumed that a sample of red cells compatible with the mother’s serum will not cause any immediate untoward effects if transfused to the infant. For example, if the mother is group B and the infant group O, group B red cells transfused at the time of birth will usually be compatible with the infant’s serum and are expected to have a normal survival (see cases reported by Jervell 1924; Wiener 1943, p. 74; Pickles 1949; first edition, p. 241).
Although the mother’s red cells are almost always compatible with her infant’s serum at the time of birth, they should not be used when she is RhD positive and her infant is D negative. For infants of less than 4 months old, the following procedures have been recommended. Both the infant’s and the mother’s ABO and RhD groups should be determined; the mother’s serum should be screened for irregular antibodies and a DAT should be done on the infant’s red cells. If the mother’s screening test and the infants DAT are both negative, blood of the same ABO and D groups as the infant may be issued without crossmatching (but see below for exceptions), even after repeated transfusions, provided that the ABO and D groups of the donor unit are checked before transfusion (Standard Haematology Practice 1991).
Haemolytic transfusion reactions may occur in infants with undiagnosed haemolytic disease of the newborn. For example, increased jaundice may follow the transfusion of D-positive blood to an infant with RhD haemolytic disease. There are special risks in group A or B infants born to group O mothers. In mature infants with ABO haemolytic disease, transfused adult A1 red cells are destroyed more rapidly than the infant’s own red cells and severe jaundice or even haemoglobinuria may result (see Chapter 12). In premature A or B infants born to O mothers, ABO haemolytic disease is believed not to occur because the A and B antigens are so poorly developed. On the other hand, the transfusion of group A or B red cells may be followed by hyperbilirubinaemia and haemoglobinuria, due to destruction of the donor’s red cells by passively acquired IgG anti-A or anti-B (see Chapter 12). Because of this risk, it has been recommended that in infants of group O mothers, group O red cells should be transfused unless it has been shown that the mother’s IgG anti-A or -B titre is less than 32 (Standard Haematology Practice 1991).
Cases in Which All Donors Seem Incompatible
In some circumstances it may be very difficult to find a donor whose red cells do not react with the patient’s serum in vitro.
Related posts:
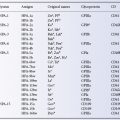 Immunology of leucocytes, platelets and plasma components
Immunology of leucocytes, platelets and plasma components
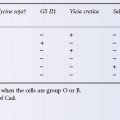 Red cell antibodies against self-antigens, bound antigens and induced antigens
Red cell antibodies against self-antigens, bound antigens and induced antigens
 Alternatives to allogeneic transfusion
Alternatives to allogeneic transfusion
 The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
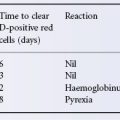 The Rh blood group system (including LW and RHAG)
The Rh blood group system (including LW and RHAG)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


