William B. Ershler
Blood Disorders in Older Adults
There is an old maxim—“Hematology is the study of blood, and the organs it perfuses.” This would seem particularly germane to geriatric medicine. Accordingly, the nuances of the interactions of blood on other organs are outside the scope of this chapter. Instead, we provide broad strokes of key topics in modern hematology while maintaining our focus on geriatric issues of clinical relevance.
Most circulating cells are derived from a single hematopoietic stem cell. The life span of blood cells is genetically predetermined, and the stem cells from which these cells are derived are programmed to survive well in excess of the human life span.1 Hematopoietic stem cells lack the surface molecules associated with differentiation (maturation), but they do express the CD34+ molecule on their surface, a feature that has important clinical implications. Hematopoietic stem cells give rise to unipotent progenitor cells that then divide and differentiate into recognizable cells of a specific lineage. This differentiation process is considered to be random. Once a specific cell line has been determined (e.g., leukocyte, erythrocyte), the progenitor cells go through further differentiation to manifest their ultimate predestined phenotype. This is accomplished by upregulation of specific receptors under the influence of multiple cytokines and growth factors. Some of these cytokines—for example, erythropoietin and granulocyte colony-stimulating factor—have, in recent years, become important clinical tools.
With aging, there is a notable reduction in the capacity to produce new blood cells. However, unless there is substantial physiologic stress, the number of circulating cells remains fairly constant. Quantitative deficiencies are only apparent when stress produces a demand that exceeds the reserve proliferative capacity. This might occur during an acute infection or after cytotoxic chemotherapy. Significant qualitative deficiencies in blood cell function are not thought to be a consequence of aging, but may be associated with age-associated chronic diseases.2
Acknowledging that the discipline of hematology extends well beyond the scope of this chapter, we attempt in this chapter to provide an overview of blood disorders particularly relevant to older adults and common enough to be encountered by the practicing geriatrician. These include anemia, myelodysplasia, myeloproliferative disorders, hematologic malignancies, and disorders of hemostasis.
Anemia
Anemia is a significant health problem in older adults due to a high prevalence and significant associated morbidity (Table 91-1).3 Although there has been a long-established perspective that anemia in older adults is of little consequence, studies have shown that even mild decreases in hemoglobin levels are associated with reduced quality of life, clinical depression, falls, functional impairment, slower walking speed, reduced grip strength, loss of mobility, worsening comorbidities, mortality, and anemia. These therefore require a greater focus on this problem.4 Currently, screening for anemia is not generally practiced, and thus it is typically discovered during a workup for other conditions, when many of its deleterious effects may have already occurred.
TABLE 91-1
Anemia Prevalence in Older Adults Using the WHO Criteria
| Study | Age (yr) | Population | Prevalence (%) |
| Gurlanik15 | ≥65 | Community-dwelling older Americans | 10.6 |
| Ferrucci281 | >70 | Community-dwelling older Italians | 11 |
| Denny282 | ≥71 | Community-dwelling older adults | 24 |
| Joosten283 | ≥65 | Hospitalized older adults | 24 (defined as hemoglobin < 11.5 g/dL) |
| Artz16 | Most ≥ 65 | Nursing home residents | 48 |
| Robinson19 | Most ≥ 65 | Nursing home residents | 53 |
| Sahin20 | Most ≥ 65 | Nursing home residents | 54 |

The World Health Organization (WHO) has defined anemia as a hemoglobin level of less than 13 g/dL in adult males and less than 12 g/dL in adult females.5 In older men and women, anemia by this definition is associated with an increase in mortality.6–11 It has been noted that the WHO criteria do not take into account inherent racial variations, particularly with respect to African Americans, who may have lower levels of hemoglobin without significant adverse outcomes.12,13 In a study that analyzed 1018 black and 1583 white older adults aged 71 to 82 years, anemia was associated with increased mortality in whites but not blacks.12,13
The issue of defining criteria for the diagnosis of anemia is also relevant in the context of age. Older women, for example, have better physical performance and function at hemoglobin values between 13 and 15 g/dL than from 12 to 12.9 g/dL,14 suggesting perhaps that a cutoff level of 12 g/dL is too low. Nevertheless the WHO definition remains the current standard used in most current epidemiologic surveys and many clinical laboratories.
Prevalence of Anemia
Guralnik and colleagues have examined the third National Health and Nutrition Examination Survey (NHANES III) database, a nationally representative sample of community-dwelling individuals; they determined age- and gender-specific prevalence rates of anemia in the total U.S. population.15 For those older than 65 years, by WHO criteria, approximately 11% were anemic (see Table 91-1). The prevalence of anemia was lowest (1.5%) among males from 17 to 49 years of age and highest (26.1%) in men older than 85 years. Among those 65 years of age and older, the prevalence rate was notably higher in African Americans compared to whites and Hispanics. Prevalence rates of anemia in older adults vary in community-dwelling and institutionalized populations. It is also clear that anemia is more common among frail older adults. In nursing homes, for example, anemia prevalence approaches 50% or higher.16–20
Pathogenesis
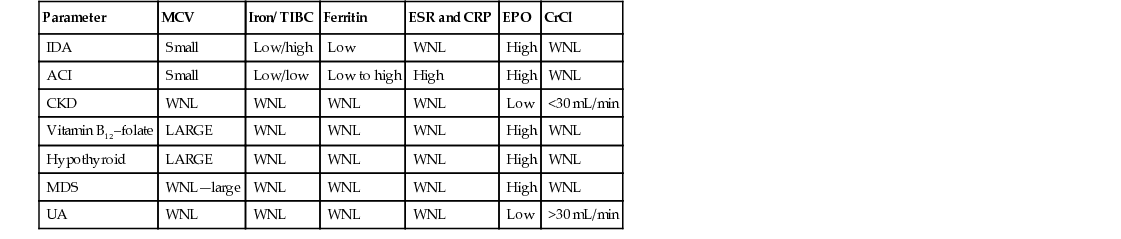
In younger adults with anemia, the cause is usually readily apparent. In older adult patients, however, discerning the cause of anemia can often be challenging (Table 91-2). Inflammation, nutritional deficiencies, and renal insufficiency are commonly observed, but for as many as one third of anemic older adults (and almost 50% for those residing in nursing homes), the anemia cannot be attributed to conventional causes, a condition now termed unexplained anemia.21
Anemia of Inflammation
Chronic diseases, such as atherosclerosis, diabetes, arthritis, infection, and malignancy, increase in prevalence with age, and each is characterized by inflammatory processes.22 Although there are several ways in which inflammation can negatively influence erythropoiesis, disordered iron kinetics is a common feature.23–25 During inflammation, there is reduced iron absorption from the gastrointestinal (GI) tract and defective reutilization of iron sequestered in reticuloendothelial cells. Hepcidin, a 25-amino-acid polypeptide produced in the liver in response to inflammatory stimuli, downregulates intestinal iron absorption and macrophage and monocyte iron release, thereby creating functional iron deficiency and resultant hypoproliferative anemia.26–30 Reduced secretion of erythropoietin due to the action of inflammatory cytokines is also known to play a role in the anemia of chronic inflammation.31,32
Iron Deficiency Anemia
The prevalence of iron deficiency in older adults ranges from 2.5% to as high as 30% in some studies. Iron deficiency is usually secondary to iron loss, rather than inadequate intake, and is important to identify because it may be a manifestation of an occult malignancy.33 For example, in one study of 114 outpatients referred to a gastroenterologist for investigation of iron deficiency, 45 had upper GI bleeding and 18 had colonic sources of bleeding.34 In an older study of 100 patients in whom the site of bleeding could not be established by any means other than laparotomy, a malignancy was found to be the cause in 10%.35 In a survey of 1388 patients older than 65 years, 25% were anemic, and approximately one third were iron-deficient. Of those with iron deficiency, GI endoscopy found 57% to have an upper GI lesion and 27% to have colonic lesions. In total, GI malignancy was found in 15% of those with iron deficiency anemia.36
Iron deficiency may be the result of GI malabsorption, particularly in patients who have had bowel resection or inflammatory bowel disease or have been on chronic antacid therapy. Furthermore, malabsorption of iron may an early manifestation of celiac disease.37,38
For patients who present with anemia and microcytic red blood cell indices, serum levels of iron, ferritin, and transferrin saturation are typically low, and the total iron-binding capacity is elevated. The coexistence of chronic inflammatory disease may complicate analysis. To determine whether patients with chronic inflammation and anemia are iron-deficient, measurement of the soluble transferrin receptor is frequently useful. Under conditions of iron deficiency, the transferrin receptor is upregulated, and increased levels are found in the serum. An index derived by dividing the serum level of soluble transferrin receptor by the log of the ferritin level, has been shown to be helpful.39 A ratio less than 2 denotes anemia of chronic inflammation, whereas a value greater than 2 identifies patients with uncomplicated iron deficiency anemia or a combination of iron deficiency and inflammation.
In light of the adverse consequences of anemia, older patients with anemia should be treated as possible prior to diagnostic studies to define the cause for the iron deficiency. Parenteral iron is commonly prescribed in current practice,24 but it is notable that oral replacement can be very effective, even at low doses.40
Vitamin B12 and Folate
Since the implementation of the U.S. Food and Drug Administration (FDA) policy of folic acid fortification of cereal grain products in 1998, there has been a dramatic reduction in measurable folate deficiency. Just 2 years after implementation, examination of the NHANES cohort IV (1999 to 2000), compared with cohort III (1988 to 1994) revealed the prevalence of low serum folate concentrations (<6.8 nmol/L), decreased from 16% before to 0.5% after fortification.41 Thus, folic acid deficiency is currently an uncommon cause of anemia. Vitamin B12 deficiency, however, remains a problem, particularly in older adults.42 Most cases of vitamin B12 deficiency in older adults is due to food cobalamin malabsorption, with true dietary deficiency or pernicious anemia being significantly less common. An age-associated atrophic gastritis, with or without antacid therapy, is a frequent antecedent. Macrocytic indices, the hallmark of vitamin B12 deficiency, may not be apparent because of concomitant inflammatory disease or iron deficiency.
In addition to red cell changes, patients with vitamin B12 deficiency may also present with a myriad of hematologic abnormalities, including leukopenia, thrombocytopenia, and pancytopenia, occasionally requiring bone marrow to distinguish it from myelodysplasia or aplastic anemia. Vitamin B12 deficiency is usually suspected by the finding of an elevated mean corpuscular volume (MCV) on routine screening or during evaluation of the cause of the anemia. However, other causes for macrocytosis are more common; these include excessive alcohol intake, drug intake (particularly antineoplastic agents), reticulocytosis, myelodysplasia, and hypothyroidism. Levels of vitamin B12 below 200 pg/mL reliably indicates vitamin B12 deficiency; however, measurement of methylmalonic acid and homocysteine levels, which are elevated, may be necessary to establish the diagnosis for those with higher serum vitamin B12 levels. Treatment with intramuscular vitamin B12 and crystalline oral vitamin B12 are effective in older adults, even in patients with cobalamin food malabsorption.43
Renal Insufficiency
Chronic kidney disease (CKD) is an important cause of anemia in older adults.44,45 Reduced renal erythropoietin (EPO) production is the primary factory leading to anemia in CKD. Serum EPO levels have been shown to be inappropriately low at a creatinine clearance of less than 40 mL/min.46 The precise degree of renal dysfunction sufficient to cause anemia remains controversial. Mild hemoglobin decreases in adults may be detected at a creatinine clearance of 40 to 60 mL/min.47,48
A survey of community-dwelling older adults suggested anemia and low EPO levels independent of age and other factors at a creatinine clearance less than 30 mL/min.49 Of 6200 residents in a nursing home, 59% were found to be anemic and 43% had calculated creatinine clearances of less than 60 mL/min. This analysis indicated that renal impairment, even at mild levels, increases the risk of anemia.19
Unexplained Anemia
Increasingly, it has become recognized that approximately one third of older adults with anemia do not have an obviously discernible cause on routine evaluation (Table 91-3). Typically, this anemia is generally mild (hemoglobin concentration in the 10- to 12-g/dL range), normocytic, and hypoproliferative (low reticulocyte count). It has been postulated that the cause relates to a number of factors, including testosterone level,50 occult inflammation,51 reduced hematopoietic reserve with advancing age,52 inappropriately low serum EPO level,53,54 and myelodysplastic syndrome (see later). It is clear that this anemia is associated with a low serum EPO level for the degree of anemia. The EPO level usually falls within the normal reference range. However, this is abnormal, because the serum EPO level should increase with falling hemoglobin concentration. The diagnosis of unexplained anemia assumes that the clinician has excluded serious causes. The threshold for performing a bone marrow examination to exclude myelodysplastic syndromes remains unknown. However, we advocate considering a bone marrow examination in all patients requiring red cell transfusion who otherwise have an unexplained anemia. Macrocytosis, thrombocytopenia, neutropenia, splenomegaly, or unexplained constitutional symptoms of fever, chills, early satiety, bone pain, and/or weight loss should prompt consideration of a bone marrow examination.
TABLE 91-3
Features of Unexplained Anemia
| Parameter | Value |
| Hemoglobin | 10.5-12 g/dL |
| Reticulocyte index | Low |
| MCV | 80-95 fL |
| Serum Iron | Mildly low normal |
| TIBC | Normal |
| Iron saturation (%) | Mildly low normal |
| Vitamin B12, folate, ESR, TSH | Normal |
| Platelet and white blood counts | Normal |
| Creatinine clearance | <90 to >30 mL/min |
Myelodysplasia
The term myelodysplastic syndrome (MDS) includes a heterogeneous group of disorders characterized by dysplastic changes within the bone marrow and impaired proliferation in one or more cell lines (e.g., erythroid, myeloid, megakaryocytic).55 As such, peripheral cytopenias and symptoms related to anemia, leukopenia, or thrombocytopenia are common manifestations of the disease. MDS occurs primarily in older men, with median age of 76 years. Each year, approximately 15,000 patients will be diagnosed with this disorder, although this might be an underestimate.56 The continues to rise, with the highest incidence found in those older than 80 years (>30 cases/100,000/year).57–59
Classification
The long-established French-American-British (FAB) classification60 has defined five distinct type of MDS—refractory anemia (RA), refractory anemia with ringed sideroblasts (RARS), refractory anemia with excess blasts (RAEB), refractory anemia with excess blasts in transformation (RAEB-T), and chronic myelomonocytic leukemia (CMML). This general classification has been modified by WHO based on salient bone marrow cytogenetic features (e.g., deletion of the short arm of the fifth chromosome [5q-]) and other histopathologic or clinical features (Table 91-4).61 Thus, RAEB is divided into two groups based on the number of blasts and RA and RARS are divided into those with only anemia or those with multilineage involvement. These modifications have enabled a more accurate estimation of prognosis.62
TABLE 91-4
WHO Classification and Criteria for the Myelodysplastic Syndromes
| Disease | Blood Findings | Bone Marrow Findings |
| Refractory anemia (RA) | Anemia | Erythroid dysplasia only <5% blasts <15% ringed sideroblasts |
| Refractory anemia with ringed sideroblasts (RARS) | Anemia | Erythroid dysplasia only <5% blasts ≥15% ringed sideroblasts |
| Refractory cytopenia with multilineage dysplasia (RCMD) | Cytopenias (bicytopenia or pancytopenia) No or rare blasts No Auer rods <1 × 109/L monocytes | Dysplasia in ≥10% of cells in two or more myeloid cell lines <5% blasts in marrow No Auer rods <15% ringed sideroblasts |
| Refractory cytopenia with multilineage dysplasia and ringed sideroblasts (RCMD-RS) | Cytopenias (bicytopenia or pancytopenia) No or rare blasts No Auer rods <1 × 109/L monocytes | Dysplasia in ≥10% of cells in two or more myeloid cell lines ≥15% ringed sideroblasts <5% blasts No Auer rods |
| Refractory anemia with excess blasts-1 (RAEB-1) | Cytopenias <5% blasts No Auer rods <1 ×109/L monocytes | Unilineage or multilineage dysplasia 5% to 9% blasts No Auer rods |
| Refractory anemia with excess blasts-2 (RAEB-2) | Cytopenias 5% to 19% blasts With or without Auer rods <1 × 109/L monocytes | Unilineage or multilineage dysplasia 10% to 19% blasts With or without Auer rods |
| Myelodysplastic syndrome, unclassified (MDS-U) | Cytopenias No or rare blasts No Auer rods | Unilineage dysplasia in granulocytes or megakaryocytes <5% blasts No Auer rods |
| MDS associated with isolated del(5q) | Anemia <5% blasts Platelets normal or increased | Normal to increased megakaryocytes, with hypolobated nuclei <5% blasts No Auer rods Isolated del(5q) |
Pathogenesis
Although no distinct cause for MDS can be identified in most cases, there is an association with prior chemotherapy in many cases, particularly with alkylating agents or topoisomerase inhibitors and with prior radiation therapy.63,64 Treatment-related MDS patients are usually younger and have a worse prognosis than de novo MDS patients.63 Prolonged exposure to high concentrations of benzene and pesticides appears to increase the risk of MDS, possibly by inducing chromosomal abnormalities.65 Carcinogens present in cigarette smoke appear to have a similar effect in increasing the risk of MDS, especially of the type associated with deletions of chromosomes 5 and 7.66 A familial cause for MDS has also been postulated.65 Chromosomal abnormalities are frequently seen in MDS, very similar to those in acute myelogenous leukemia (AML), including complex karyotypes.63 Several mechanisms may be involved in the pathogenesis of MDS, including alterations in apoptotic pathways, cytokine regulation, bone marrow microenvironment, mitochondrial enzymes, and immune regulation.67 Furthermore, there has been evolving evidence that some MDS cases have an autoimmune basis because, clinically, MDS may be associated with other autoimmune disorders, and laboratory evidence has documented oligoclonal T cell patterns in up to 50% of cases.68
Clinical and Laboratory Features
Patients with MDS may be asymptomatic or have symptoms and signs related to qualitative or quantitative defects of erythrocytes, leukocytes, and platelets.69 Fatigue, which significantly affects quality of life,70 exertional dyspnea, fever, and infections, are some of the reasons for consulting a physician. A complete blood count usually reveals anemia, with normocytic or macrocytic indices. Moderate leukopenia and thrombocytopenia or thrombocytosis may be present. A bone marrow examination may reveal a normal or hypercellular pattern, with dysplastic features identified in erythroid, myeloid, and megakaryocytic cell lines or in all three (trilinear). Commonly seen changes in peripheral blood and bone marrow include anisocytosis, macrocytosis, basophilic stippling, ringed sideroblasts, pseudo–Pelger-Huet abnormality, blast cells, and hypersegmented neutrophils, micromegakaryocytes, and large platelets.69
Prognosis
The International Prognostic Scoring System (IPSS)71,72 divides patients into low, intermediate-1 (INT-1), intermediate-2 (INT-2), and high categories (Table 91-5). It is based on the number of cytopenias (hemoglobin level < 10 g/dL, absolute neutrophil count < 1,500/µL, platelet count < 100,000/µL), chromosomal abnormalities, and percentage of bone marrow blasts and is used to estimate overall survival and predict progression to AML. Median survival ranges from less than 3 months, regardless of age, in the high-risk groups to almost 12 years in those younger than 60 years in the low-risk group. In the low-risk group, patients older than 70 years seem to have a worse prognosis than younger patients, but age does not seem to change the prognosis significantly in those in the high-risk category. Other factors found to be of prognostic importance include WHO subtypes73 and transfusion dependence. The IPSS and Revised IPSS (IPSS-R) are used to predict the prognosis of patients with MDS at the time of diagnosis.71,72 Management recommendations for patients with MDS have largely been based on their IPSS score.74 Low-risk MDS includes patients with an IPSS score of low/INT-1, and high-risk MDS includes those with an IPSS score of INT-2/high.74 Older age did not influence prognosis in the refractory anemia group, which is usually associated with good prognosis.73 Also, MDS associated with a 5q deletion is associated with a good prognosis.75

Treatment
Supportive care in the form of transfusions, iron-chelating therapy, and adequate treatment of infection is the backbone of treatment for those with MDS. In the low and INT-1 groups, treatment is deferred unless significant cytopenias are present. Patients with deletion 5q have relatively good prognosis and experience an excellent response to the thalidomide analogue lenalidomide. In one series, 67% of patients became transfusion-independent after treatment with 10 mg of daily lenalidomide and continued to remain so for more than 2 years.76 Recombinant erythropoietin, with or without granulocyte-stimulating factor, is used to treat symptomatic anemia in those with low serum erythropoietin levels and symptomatic anemia.77 Older patients in the low INT-1 category but with neutropenia or thrombocytopenia and patients in the INT-2 and high-risk group who are ineligible for transplantation are best treated with DNA methyltransferase inhibitors, such as decitabine78 or azacytidine79; both have been shown to improve quality of life. Although intensive therapy with allogeneic stem cell transplantation has been traditionally reserved for younger patients, reduced-intensity conditioning regimens have extended this therapy to selected older patients, with favorable results. Assessment with the hematopoietic cell transplantation comorbidity index (HCT-CI) has been used in older MDS patients and may help select patients for whom the risk-benefit ratio is favorable for this form of aggressive therapy.80
Myeloproliferative Disorders
Myeloproliferative disorders (MPDs) are characterized by clonal proliferation of hematopoietic stem cells. They include polycythemia vera (PV), essential thrombocythemia (ET), chronic idiopathic myelofibrosis (CIF), and chronic myelogenous leukemia (CML). CML is identified by the presence of the translocated bcr-abl gene and typically has three phases—a chronic phase, seen in more than 80% of patients, blastic phase, and intermediate accelerated phase.
The new 2008 WHO classification has replaced the term disease in MPD with neoplasm to indicate the possibly malignant nature of these disorders.61,81 The myeloproliferative neoplasm (MPN) group includes mast cell disease, chronic eosinophilic and neutrophilic leukemia, hypereosinophilic syndrome, and other previously unclassified bone marrow disorders in addition to CML, PV, CIF, and ET. The common feature of these disorders is evident clonal proliferation without dysplasia.
Epidemiology
PV and CIF are more often diagnosed in older adults,56 equally in both genders, with a median age of 70 years. Incidence rates for all age groups have ranged from 0.7 to 2.6/100,000 for PV and 0.3 to 1.5/100,000 in CIF, but rates as high as 23.5/100,000 have been reported for those from 70 to 79 years of age for PV. An increased incidence among Ashkenazi Jews has been reported. Incidence for ET ranges from 0.59 to 2.53/100,000, with almost twofold higher rates seen in women than in men. Unlike PV, ET is diagnosed at a younger age and is often seen in association with pregnancy. A significant trend toward an increase in ET incidence has been observed, especially among men, although the trends for PV and CIF have not shown any change.56 Familial studies have shown an almost fivefold increased risk for PV and sevenfold risk for ET in first-degree relatives with MPDs.82
Causative Factors
Identification of a specific mutation (V617F) in the Janus Kinase-2 gene (JAK2) has improved our understanding of the pathogenesis of myeloproliferative disease.83 JAK2 V617F can be identified in almost 95% of PV patients84 and in more than 50% of ET and CIF patients.85 Increased activity of this gene enhances the sensitivity of the mutated stem cells to hematopoietic growth factors such as erythropoietin, thrombopoietin, stem cell factor, and granulocyte-stimulating factor, causing clonal trilinear proliferation of myeloid precursors. Also, a mutation in the mpl gene, which involves the thrombopoietin receptor, is also thought to play a minor role in the pathogenesis.86 The bcr-abl fusion gene formed as a result of translocation of the abl gene from chromosome 9 to the bcr region on chromosome 22 causes transcription of proteins with abnormal tyrosine kinase activity, resulting in clonal proliferation usually recognized in CML.87 Clonal evolution, with additional chromosomal abnormalities frequently involving the 17th, are seen in more than 50% of patients in an accelerated phase of the disease.88
Production of growth factors by cytokines secreted from megakaryocytes and monocytes are hypothesized to play a role in the proliferation of fibroblasts, changes in the extracellular matrix, and angiogenesis seen in CIF.89,90 Abnormal homing of stem cells and endothelial progenitor cells CD34+ to peripheral hematopoietic organs are also known to play a role in the extramedullary hematopiesis of myelofibrosis.91 Secondary myelofibrosis can result because of the progression of PV and ET, although the progression of true ET has been highly debated.
Clinical Features and Diagnosis
The characteristic clinical features vary by the specific MPD. In ET, they are related to abnormalities in platelet number and function; this is manifest predominantly as thrombotic episodes, although hemorrhagic episodes are also encountered.92 Thrombotic events can precede the diagnosis of the disease and can involve microvascular and large-vessel arterial circulation, causing ischemic strokes, peripheral vascular disease, and myocardial infarction in addition to venous thrombosis.93 Erythromelalgia, a specific microvascular condition seen in ET, typically presents with erythremic burning sensations of the extremities and ulceration of the toes.94 The risk of thrombosis increases with age in PV and ET.95 A higher incidence of cardiovascular complications is also seen in those older than 65 years who have had a history of thrombosis.96 It appears that the presence of a JAK2 mutation92 and leukocytosis97 may be additional risk factors for thrombosis in MPN patients. Hemorrhagic complications usually occur in patients with very high platelet counts (>1500 × 109/L); the clinical presentation can vary from minor bruising or epistaxis to life-threatening GI bleeding.95 CIF may be asymptomatic in more than 30% of patients; the diagnosis may be suspected on the basis of a leukoerythroblastic blood picture, presence of classic teardrop cells, or an enlarged spleen, usually in the prefibrotic stage.98 Presentation in CML is similar to that seen in other MPNs, with constitutional symptoms and hemorrhagic manifestations. A finding of a low leukocyte alkaline phosphatase (LAP) level characterizes CML as compared to the PV, ET, and CIF, which that typically have a normal LAP level.
General signs and symptoms in patients with MPDs include splenomegaly, which can run the spectrum of mild and of little concern to uncomfortable and even life-threatening if rupture occurs. Pruritus may be present in more than 50% of patients. Patients with MPD also have a high incidence of fatigue, which may substantially decrease the quality of life.99 Weight loss, night sweats, fever, and bone pain are also seen.
Diagnosis
The diagnosis of MPN has undergone a radical change with identification of the JAK 2 mutation. Previously, standard criteria included physical signs and parameters based on peripheral blood counts, oxygen saturation, bone marrow findings, red cell mass, and presence of splenomegaly. The new WHO criteria, an update of the previous 2001 criteria, include detection of a JAK2 mutation along with bone marrow biopsy in the diagnosis of bcr-abl–negative MPNs. Important changes also include lowering the platelet threshold number for the diagnosis of ET from 600 to 450 × 109/L and establishing a 2-g/dL increase of hemoglobin from normal levels as criteria for PV.100 Different algorithms using serum EPO levels, morphology of bone marrow megakaryocytes, bone marrow reticulin staining, and use of fluorescence in situ hybridization (FISH) for bcr-abl have been proposed to enable differentiation of one MPN from another.100 It is especially important to differentiate the cause of isolated thrombocytosis, because the prognosis of ET is very different from the thrombocytosis seen in early prefibrotic CIF and that due to CML. The diagnosis of ET also may require exclusion of reactive thrombocytoses such as cancer, iron deficiency anemia, and inflammatory conditions.101 The diagnosis of CML is straightforward via identifying the Philadelphia chromosome by cytogenetics or the bcr-abl gene by the FISH technique.
Prognosis
As noted, survival times vary markedly in each of the MPDs described. One study has shown a 3-year survival rate as high as 92% for ET and 88% for PV. However, older age was associated with lower overall survival for MPDs as a group (90% in those < 50 years and 66% in those > 80 years).56 ET and PV are associated with reduction in life expectancy, significant morbidity related to thrombosis, hemorrhage, and a small but definite risk for leukemogenic transformation or marrow fibrosis, especially in older adults.102 Fatigue present in more than 80% of these patients has significant negative effect on the quality of life.99 It is now recognized that those with the JAK2 mutation,92 especially those with higher allelic burden and higher leukocyte counts, have a tendency to higher rates of thrombosis. Age older than 60 years, previous history of thrombosis,96 and high platelet count (>1500 × 109/L) were previously identified as important prognostic factors for thrombosis in these patients. Patients in the prefibrotic stage of CIF have a relatively good prognosis, with a survival of almost 12 years. However, for those with high-risk CIF, life expectancy is significantly reduced.103 The median length of survival can be as short as 1 year for those at highest risk. AML in patients with primary myelofibrosis carries the worst prognosis, with a dismal median survival of less than 3 months. Numerous scoring systems in CIF using hemoglobin levels, age, and white blood cell count have been described to predict prognosis,104 and older age appears to be a common negative prognostic factor.105
Treatment
Treatment in PV and ET is generally recommended to reduce the morbidity associated with qualitative and quantitative changes in blood cells. Phlebotomy to reduce the hematocrit to less than 45% is the mainstay of treatment in PV because this has reduced the incidence of thrombosis.106 Low-dose aspirin has been shown to reduce the risk of microvascular and macrovascular complications and is recommended for treatment of all patients with PV.107 In the European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study, which randomized predominantly asymptomatic low-risk PV patients, the incidence of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, and major venous thromboembolism was lower among those treated with 100 mg aspirin daily as compared to placebo-treated controls (relative risk [RR], 0.4; 95% confidence interval [CI], 0.18 to 0.91]; P = .0277).108 Total and cardiovascular mortality were also reduced by 46% and 59%, respectively. There was no significant increase in the risk of bleeding. In younger low-risk ET patients, treatment is generally not required, although low-dose aspirin can be used to reduce microvascular symptoms.109 However, in older patients, aspirin may be recommended based on the ECLAP study, especially if other traditional risk factors for vascular disease are also present.109
The evidence for benefit with treatment with hydroxyurea (HU) in high-risk ET patients has been clearly demonstrated, particularly with reference to reduced thrombosis (1.6%) compared to no treatment (10.6%).110–112 Superior benefit was also seen when HU was compared to other cytoreductive agents, including anagrelide.110 Studies with HU in PV have not been as consistent, and a higher risk of leukemic transformation has been observed in treated patients. However, in high-risk PV and ET patients, HU is considered the standard of care.106,112
Primary myelofibrosis is an intractable illness, refractory to standard approaches, and allogeneic hematopoietic stem cell transplantation (HSCT) remains the only potentially curative therapy available. However, age and increasing comorbidity are strong negative prognostic factors for survival, even in these patients, and only 14% of MF transplantation patients older than 45 years survived 5 years after allogeneic HSCT as compared to 62% of younger patients.113 In that same series, those with a Charlson comorbidity index of 4 to 6 had a greater than twofold risk of mortality. Reduced-intensity conditioning (RIC) regimens have increased response rates and event-free survival (EFS) in older patients but, in most of these studies, patients were considered “old” at the age of 60 years. For patients younger than 70 years, with low comorbidity, an RIC regimen and allogeneic transplantation are reasonable options, worthy of clinical investigation.114,115 Optimum treatment for those 70 years of age and older remains investigational and, outside of a clinical trial, may be supportive management alone. Control of symptomatic anemia and elevated leukocytes or platelets may require pharmacologic intervention, but treatment is primarily palliative. Splenomegaly is a classic feature of MF, and splenic radiation has provided effective palliation in selected cases.116
Erythropoietin and danazol were commonly used to correct anemia, but a recent retrospective study117 has found an increased risk of transformation to leukemia with the use of these agents in myelofibrosis; thus, enthusiasm for this approach has been tempered. High response rates for patients with anemia and splenomegaly with lenalidomide have been observed in those with the del(5q) group of myelofibrosis.118 However, patients with del(5q) are exceedingly uncommon.119
The management of CML has changed dramatically in the past 2 decades. Survival in CML patients depends on the phase of the disease, with median survival now measured in decades for those presenting in the chronic phase,120 but still less than 3 months for those in the blastic phase.121 Prognostic classification incorporating variables such as percentage of blasts, spleen size, platelet count, basophil and eosinophil counts, and age distinguishes three groups—low, intermediate, and high risk—with good correlation with survival.122 With the introduction of the oral tyrosine kinase inhibitor imatinib and, more recently, with the second-generation tyrosine inhibitors dasatinib123 and nilotinib,124 treatment of CML has improved considerably; it appears that response rates and overall survival for CML patients approach that of age-matched individuals without CML.120 The good news is that older patients respond well to tyrosine kinase therapy, with responses comparable to those observed for younger patients.125,126
Other Hematologic Malignancies Common to Older Patients
Multiple Myeloma
Multiple myeloma (MM), the second most common hematologic malignancy diagnosed in the United States, is a disorder characterized by monoclonal proliferation of plasma cells (>10%) within the bone marrow. Typically, in MM, abnormal amounts of monoclonal immunoglobulin or light chains (kappa or lambda) are present in the serum and/or urine. Manifestations related to involvement of the bone predominate, but symptoms resulting from the extramedullary effects of the circulating abnormal protein or hypercalcemia are also frequently encountered. The median age of myeloma diagnosis is 70 years, and it occurs more frequently in African Americans and in males. The U.S. annual incidence is 4.4/100,000. Curiously, MM incidence and mortality rates are decreasing. However, the 5-year relative survival rates continue to remain less than 20% for patients 65 years and older.127
Causative Factors
Lower socioeconomic status, education, and dietary habits seem to account for the differences in incidence between African Americans and whites in the United States.128,129 Familial studies have shown that the risk of myeloma is higher in patients with a family history of hematologic malignancy, possibly implicating a genetic predisposition.130 Risk is also known to be higher after radiation or exposure to pesticides, certain chemicals, and asbestos131 and also in those with a history of inflammatory disease132 or allergies.133 High levels of human herpesvirus 8 (HHV-8) virus have been reported in bone marrow stromal cells of myeloma patients, indicating a possible viral cause for the disease in selected cases.134
Pathogenesis
Bone pain is the primary symptom in approximately 70% of patients. Radiographs reveal localized punched-out lytic bone lesions or diffuse osteoporosis, usually in bones with active hematopoietic tissue. The discrete lytic lesions are characterized by large and numerous osteoclasts on the bone-reabsorbing surface. An excess of osteoclast-activating factors such as receptor activator of nuclear factor-kappa B ligand (RANKL), macrophage inflammatory protein-1α (MIP-1α), and interleukin-6 (IL-6), coupled with a decrease in osteoblast activity due to the presence of inhibitors such as Dickkopf-related protein 1(DKK-1), contribute to the characteristic osteolytic bone lesions. With bone demineralization and decreased activity because of pain, hypercalcemia may be expected. The symptoms of hypercalcemia (drowsiness, confusion, nausea, thirst) are nonspecific, but their occurrence should alert the physician to investigate this possibility. Cardiac arrhythmias, renal insufficiency, and profound central nervous system (CNS) depression can develop as hypercalcemia progresses.
High levels of circulating monoclonal protein can cause increased whole blood viscosity. This occurs most frequently with immunoglobulin M (IgM; see Waldenstrom disease, later), but may also occur with IgA or even IgG myeloma. Increased viscosity compromises circulation, including that to the brain, kidney, and heart, and symptoms of confusion, light-headedness, and/or chest pain may result. The myeloma protein itself may cause tubular damage in the kidney, and renal insufficiency is a major consequence of uncontrolled myeloma.
Clinical Features
It is important to differentiate myeloma from other plasma cell dyscrasias that occur with increasing frequency with age. Monoclonal gammopathy of undetermined significance (MGUS) once considered a benign disorder secondary to age-associated immune dysregulation,135 is now thought to be a precursor to myeloma, with a conversion of MGUS to MM of about 1%/year.136,137 The features of MGUS (Table 91-6) include a marrow with less than 10% plasma cells (and without dysplastic features characteristic of myeloma plasma cells), less than 3-g/dL monoclonal protein in the serum, and no end-organ damage. MGUS patients who have more than 1.5 g/dL of monoclonal protein other than IgG and an abnormal urinary light chain ratio are at a greater risk for earlier progression to MM. Follow-up evaluation, including serum protein electrophoresis and quantitation of the monoclonal protein, is recommended every 3 months so that early conversion to myeloma may be recognized.138
TABLE 91-6
Early Myeloma versus Monoclonal Gammopathy of Undetermined Significance (MGUS)
| Parameter | Early Multiple Myeloma | MGUS |
| Pathogenesis | Neoplastic plasma cell disorder (malignant) | Disordered immunoregulation or possibly benign B cell neoplasm |
| Bone marrow | Frequently > 10% plasma cells with many dysplastic features (myeloma cells) | Usually 10% normal-appearing plasma cells |
| Bone | Lytic bone lesions or diffuse osteoporosis | Usually no bone disease |
| Symptoms | Bone pain, constitutional symptoms (e.g., fatigue, weight loss), or those associated with kidney failure or hyperviscosity | Usually no symptoms |
| Serum spike | Progressively rising | Stable |
Smoldering myeloma has the same laboratory features as MM but without end-organ damage. Solitary plasmacytoma is an isolated tumor consisting of plasma cells in bones (solitary myeloma of bone) or soft tissue (extramedullary plasmacytoma), without systemic involvement.139 In 1% to 2% of patients, MM can progress to plasma cell leukemia, which is more common in younger patients, has more than 20% plasma cells, and is more aggressive than MM.
Renal dysfunction is seen in more than 20% of myeloma patients at the time of diagnosis.140 The pathogenesis of the renal disease is frequently multifactorial, with contributions from paraproteins, tubular damage, tissue deposition of light chains, amyloidosis, hyperuricemia, and infection.141–143
Pathologic fractures are also common. Fatigue related to anemia occurs in 30% to 60% of MM patients and usually occurs in those with a hemoglobin level less than 10g/dL.144 Signs of amyloidosis (present in up to 30% of myeloma patients) such as macroglossia may also be present. Although constitutional symptoms can occur because of anemia, hyperviscosity, hypercalcemia, renal insufficiency, or infection, it should be remembered that in older adults, these symptoms might be less pronounced. The clinical presentation of MM has changed over the years, with a lower percentage of patients presenting with end-organ damage. MM is usually suspected by the presence of normocytic normochromic anemia and mild renal failure or hypergammaglobulinemia,145 especially in older adults. Due to the catabolic effects of myeloma-associated cytokines, older patients may present with normocytic anemia and a drop in the serum cholesterol level, even before other signs or symptoms of the disease appear.
Laboratory Data and Diagnosis
Complete blood count, metabolic profile with liver function tests, serum lactic acid dehydrogenase (LDH) level, serum β2-microglobulin, serum free light chain assay, serum and 24-hour urine protein levels, electrophoresis, immunofixation, skeletal survey, and bone marrow are all important in the initial workup of MM patients.146 Hypercalcemia, renal insufficiency, anemia, and bony abnormalities, which are the hallmarks of end-organ damage in MM, are usually identified with these evaluations. Other laboratory abnormalities include hypoalbuminemia, elevated LDH level, β2-microglobulin, platelet, and coagulation abnormalities and rouleaux formation (stacking of red cells) apparent on the peripheral blood smear.147
Protein electrophoresis detects a monoclonal protein in approximately 80% of patients in the serum and in 75% of patients in the urine. Immunofixation (or immunoelectrophoresis) allows identification of the isotype, which is usually IgG (60%) or IgA (20%). Of the 20% without detectable monoclonal protein by serum protein electrophoresis, most will have kappa or lambda light chains detected in the urine (light chain myeloma). Only about 1% of myeloma patients will be actual nonsecretors.147 In the absence of monoclonal protein, the diagnosis of MM is established by the finding of more than 30% plasma cells in the marrow.
Prognosis
Traditionally, the Durie-Salmon staging system—predominantly clinical features and amount of paraprotein—was used to identify patients at higher risk of death because it is a good indicator of tumor burden in MM. However, the International Staging System (ISS) has been shown to provide prognostic information more accurately. This system is based on the levels of serum β2-microglobulin and albumin, as well as other measures. In one survey, in stage I or low-risk MM (serum β2-microglobulin < 3.5 g/dL; serum albumin > 3.5 g/dL), median survival was 62 months. In contrast, for stage III MM (serum β2-microglobulin > 5.5 g/dL), median survival was 29 months, and for the intermediate stage (stage II), median survival was 44 months.148 Certain cytogenetic abnormalities, including those detected by FISH, such as deletion of chromosome 13, hypoploidy, t(4;14), t(4;16), 17p−, when combined with a plasma cell labeling index greater than 3% and selected laboratory data (LDH and C-reactive protein [CRP]), have been shown to improve prognostic accuracy.149
Although most prior studies did not distinguish significant differences in clinical features of myeloma in younger versus older patients, two reports are notable. The first compared those older and younger than 70 years, and the second compared those older than and younger than 50 years. Both studies showed that older patients presented with more advanced stage, poorer performance, and more adverse clinical features, including lower hemoglobin and higher creatinine levels and, in both reports, the response rates and survival were lower for those in the older group.127,150,151
Treatment
Older patients with MGUS and smoldering or asymptomatic myeloma need careful follow-up to detect and intervene if there is evidence for progression to more aggressive disease. In patients with symptomatic myeloma, treatment includes two components—one to manage the disease and the other to provide supportive treatment to control complications or end-organ damage. In older patients, treatment is especially difficult because improvements in progression-free survival and overall survival may lead to adverse effects, such as increased toxicity and poor quality of life. Proper selection of patients using geriatric assessment may reduce the possibility of these adverse outcomes.127
Previously, vincristine, doxorubicin, and dexamethasone were used as initial aggressive therapy, although it was difficult to prove that this was more effective than oral melphalan and prednisone (MP) in terms of overall survival. Thalidomide and dexamethasone have become first-line therapy and are even used as induction therapy prior to transplantation. Bortezomib, a reversible inhibitor of 26S proteosome, alone or in combination, has also been used for MM, with excellent results.152 Retrospective studies have shown similar toxicity profiles and survival outcomes in younger and older patients, despite older adults receiving reduced chemotherapy doses.153
Currently, the standard of care in older myeloma patients is treatment with melphalan, prednisone, and thalidomide (MPT). In patients between 65 and 75 years of age, median overall survival was 51.6 months for MPT compared to 33.2 months for MP and 38.3 months for autologous transplantation with a reduced-intensity melphalan conditioning regimen.154 The complications from MPT include an increased risk of venous thromboembolism, especially when administered with high-dose dexamethasone and erythropoiesis-stimulating agents, as well as peripheral neuropathy, infection, and constipation. Other combinations that have shown benefit in older patients include bortezomib or lenalidomide in combination with dexamethasone. Objective response rates as high as 90% have been seen with both these regimens.155,156 There have been several new drugs approved for the treatment of myeloma,157 although the sequence of their use and drug combinations have yet to be established. There remains some controversy with regard to maintenance therapy, although current data would suggest efficacy in terms of progression-free survival.158
Treatment with bisphosphonates have benefit with regard to lytic bone disease, bone pain, and osteopenia.159,160 Radiation is used as a palliative measure to reduce bone pain and, along with dexamethasone, to stabilize tumors of the spine to prevent spinal cord compression and fractures. Newer surgical techniques such as vertebroplasty and kyphoplasty have improved the care of patients with bony complications in MM. Appropriate use of erythropoietic agents for management of anemia, aggressive management of infections, including prophylactic antibiotics, immunoglobulin use, and treatment of hyperviscosity and hypercalcemia are all important for reducing morbidity in MM patients.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


