Biology of Treatment-Induced Mucositis
Rachel J. Gibson
Joanne M. Bowen
Emma H. Bateman
Dorothy M. K. Keefe
INTRODUCTION
This chapter is dedicated to exploring the biology of treatment-induced alimentary tract (AT) mucositis. AT mucositis is a significant complication in the clinical oncology setting. It occurs in a large percentage of patients undergoing cytotoxic chemotherapy and radiotherapy. One of the key problems with AT mucositis is that the underlying mechanisms behind its development are not entirely understood, making it extremely difficult to develop effective interventions. Animal models have provided a detailed and important source of knowledge when sampling from patients is unavailable or interventions are yet to be fully tested. This chapter focuses on how animal models have been used to facilitate our understanding of the mechanisms of AT mucositis.
PATHOBIOLOGY OF AT MUCOSITIS
AT mucositis is an extremely common toxicity occurring after chemotherapy and radiotherapy for cancer (1). AT mucositis occurs in approximately 40% of patients after standard doses of chemotherapy and in up to 100% of patients undergoing high-dose chemotherapy and hematopoietic stem cell transplantation, or radiation for head and neck cancer (2,3,4), affecting over 2 million people worldwide each year. As the name suggests, AT mucositis can occur anywhere along the alimentary canal and is associated with many symptoms, of which significant pain, ulceration, abdominal bloating, nausea and vomiting, diarrhea, and constipation are a few (5,6,7).
The potentially severe nature of AT mucositis can have some further devastating effects, including a reduction or cessation of treatment (which may decrease the chance for remission or cure) and increased stays in hospitals, leading to increased costs of treatment and use of opioids for pain management (2,3,4). In addition to the economic costs associated with AT mucositis, there is also a significant impact on the quality of life of cancer patients, with increased morbidity and mortality (8). The potential market for management interventions in mucositis is between $1 and 2 billion annually, with a number of new agents in the development pipeline looking to capitalize on this unmet need.
Historically, mucositis has been separated into oral mucositis and intestinal mucositis, both mechanistically and for applied interventions. In addition, a number of overlapping terms have been used clinically, including stomatitis, gastrointestinal toxicity, radiation esophagitis, radiation enteritis, radiation proctitis, and the radiation gastrointestinal syndrome. This lack of consistent terminology, for the underlying issue of regimen-related mucosal damage (mucositis), acts as a barrier for research and knowledge advancement. The prevailing consensus is that changes affecting the oral cavity and throat are considered oral mucositis, while changes arising lower in the digestive tract are grouped as gastrointestinal mucositis, which can include esophageal, gastric, and small and large bowel injury. A further level of contention is that gastrointestinal toxicity is sometimes cited as inclusive of the oral cavity, but not always. The concept of alimentary mucositis is relatively new and accommodates regional differences in presentation and diagnosis in an aim to facilitate a unified approach to research, each region then being a subset.
AT MUCOSITIS IN HUMANS
AT mucositis is a field that has rapidly evolved over the last 15 years. There have been exciting and significant advances leading from basic clinical science questions to conducting whole-genome microarray experiments and developing complex algorithms to uncover toxicity clustering. The first step in the AT mucositis matrix started with relatively simple investigations of pathology and malabsorption following radiation or chemotherapy in cancer patients (9,10). One example of an early study investigating the severity and time course of changes in patients’ intestinal permeability following high-dose chemotherapy and autologous blood stem cell transplantation will be discussed here (11). Briefly, this study recruited 35 patients and determined the maximum sugar permeability occurring in the small intestine 14 days after the start of chemotherapy. Levels returned to normal 7 weeks after chemotherapy (11). Furthermore, this abnormality was found to correlate with the time frame that the patients suffered from other gastrointestinal symptoms including anorexia and nausea (11). This study offered the first insight into intestinal damage after chemotherapy in patients. It led to further questions about the mechanisms by which chemotherapy damages the small intestine and led to further, more detailed clinical studies.
A second study assessed the frequency, duration, and severity of both oral and gastrointestinal symptoms following chemotherapy. Sixty patients with a variety of malignancies were recruited to this study, including newly diagnosed patients, patients undergoing high-dose chemotherapy, and patients undergoing autologous peripheral blood stem cell transplantation (12). Patients underwent a variety of tests,
including questionnaires and noninvasive nutritional assessments and the intestinal sugar permeability tests, as well as invasive blood testing for serum endotoxin. A small number of patients also underwent a series of breath tests before and after chemotherapy (12). Findings indicated that patients experienced gastrointestinal symptoms 3 to 10 days after chemotherapy commenced, with these returning to normal by day 28. In contrast to this, patients did not experience oral symptoms until day 7 after chemotherapy, remaining until day 14 before returning to normal by day 28 (12). These findings led to a third patient-based study, aimed at assessing small intestinal mucosal histology following chemotherapy. Twenty-three patients with a variety of malignancies were recruited to the study and underwent an endoscopy with duodenal biopsy before, and at varying time points after, chemotherapy. Biopsies were assessed using a variety of techniques including the apoptosis-specific assay Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL), enterocyte height, and transmission electron microscopy (13). Findings indicated a sevenfold increase in apoptosis in intestinal crypts 1 day after chemotherapy, followed by a reduction in intestinal morphometry 3 days after chemotherapy. Values had returned to normal by 16 days after chemotherapy (13). These findings are similar to the histopathological changes identified by Trier and Browning during acute phase intestinal injury induced by abdominal radiation, which described reduced mitosis, decreased thickness of the mucosa, and fragments of disintegrating epithelial cells (14).
including questionnaires and noninvasive nutritional assessments and the intestinal sugar permeability tests, as well as invasive blood testing for serum endotoxin. A small number of patients also underwent a series of breath tests before and after chemotherapy (12). Findings indicated that patients experienced gastrointestinal symptoms 3 to 10 days after chemotherapy commenced, with these returning to normal by day 28. In contrast to this, patients did not experience oral symptoms until day 7 after chemotherapy, remaining until day 14 before returning to normal by day 28 (12). These findings led to a third patient-based study, aimed at assessing small intestinal mucosal histology following chemotherapy. Twenty-three patients with a variety of malignancies were recruited to the study and underwent an endoscopy with duodenal biopsy before, and at varying time points after, chemotherapy. Biopsies were assessed using a variety of techniques including the apoptosis-specific assay Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL), enterocyte height, and transmission electron microscopy (13). Findings indicated a sevenfold increase in apoptosis in intestinal crypts 1 day after chemotherapy, followed by a reduction in intestinal morphometry 3 days after chemotherapy. Values had returned to normal by 16 days after chemotherapy (13). These findings are similar to the histopathological changes identified by Trier and Browning during acute phase intestinal injury induced by abdominal radiation, which described reduced mitosis, decreased thickness of the mucosa, and fragments of disintegrating epithelial cells (14).
A further clinical study was conducted and aimed to determine whether apoptosis also occurred in the oral mucosa following chemotherapy (14). Twenty patients with a variety of malignancies were recruited and each underwent a buccal punch biopsy prior to, and at varying time points after, chemotherapy. Biopsies were assessed using the TUNEL assay and transmission electron microscopy. Apoptosis increased in the basal layer of the buccal mucosa in the first 3 days following chemotherapy and had not returned to normal levels by 11 days after treatment. Apoptosis was accompanied by significant ultrastructural changes (14).
Each of these clinical studies clearly indicated that cancer chemotherapy causes significant damage to the AT. While each study answered a specific question, it also led to many questions that needed answers. However, in order to fully understand the mechanisms of how this damage occurs, an appropriate animal model needed to be developed. While it was possible to perform upper gastrointestinal endoscopy and biopsy in a small number of patients, more detailed investigations were required. These simply would not have been possible without the development and use of animal models.
HISTORICAL PATHOPHYSIOLOGY OF AT MUCOSITIS
AT mucositis as a field has rapidly evolved over the last decade. In 1998, Sonis first proposed that oral mucositis followed a distinct cascade of phases, namely the inflammatory/vascular; the epithelial; the ulcerative/bacteriological; and the healing phase (8). Briefly, it was initially thought that in the first inflammatory phase, following the administration of cytotoxic therapy, various proinflammatory cytokines were released from the epithelial tissues, leading to local tissue damage. This phase was postulated to occur early, within 24 hours after cytotoxic treatment. The second epithelial phase was thought to occur between 4 and 5 days following treatment. Cytotoxic agents were thought to directly inhibit deoxyribonucleic acid (DNA) replication and mucosal cellular reproduction, ultimately leading to a decrease in basal epithelial proliferation (8,15). Consequently, as not all cytotoxic drugs affect DNA synthesis (8), it was thought that there would be a higher probability that patients on particular treatment regimes would be more susceptible to oral mucositis than others. Evidence supporting this notion came from studies in children, known to have a high basal cell proliferation. These children were three times more likely to develop oral mucositis than older adults, who have a slower basal cell proliferation (16). The reduction in cellular proliferation of the oral mucosa was thought to cause mucosal atrophy, with breakdown of collagen and frank mucosal ulceration, marking the beginning of the ulcerative and bacteriological third phase. Clinically, this was the most symptomatic phase for patients and presented approximately 1 week following treatment (8). The fourth and final healing phase referred to the mucosa resuming normal basal cell proliferation and differentiation as well as controlling the oral bacterial flora. The length of this final phase was thought to be related to the duration of the condition; however, it most probably did not relate to the intensity. Additionally, anything that adversely affected wound healing would also affect this final phase (8).
CURRENT HYPOTHESIS FOR THE PATHOBIOLOGY OF AT MUCOSITIS
In 2004, it was recognized that AT mucositis was more than just an epithelial process, prompting a revision of the fourphase model to the current overlapping five-phase model (Figure 18.1). The phases were renamed as follows: 1, initiation; 2, primary damage response; 3, signal amplification; 4, ulceration; and 5, healing (17,18). In this updated model, initiation occurs immediately following exposure to cytotoxic therapy, with both DNA and non-DNA damage occurring, resulting in the generation of reactive oxygen species (ROS) and clonogenic cell death. Clonogenic cell death in the small intestine has been shown to occur within 4 hours following irradiation (19). This leads to the second phase of primary damage response. In this phase, nuclear factor kappa B (NFκB) has been shown to be a key transcription factor that is “turned on” in response to DNA damage and ROS, leading to a cascade of downstream genes including proinflammatory cytokines interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF). Radiation at physiological doses has been shown to induce both NFκB and another transcription factor, p53, in cells (20), indicating that these stress-responsive factors
are important in mucositis development. Chemotherapy treatment has shown to increase the transcription factor AP1 (21), and this family of transcription factors is known to induce damage in all cells within the mucosa and submucosa (18). In addition, increased expression of IL-1β and TNF has been detected in the submucosal cells of hamsters following acute targeted radiation (22). In the same experiment, interference with TNF-α reduced mucositis, indicating that the cytokine signaling that is set up relatively early on plays a key role in development of injury. In the third phase of signal amplification, the proinflammatory cytokines that were upregulated earlier begin to amplify the original signal and positively feed back on NFκB, thus causing further amplification. In addition, biologically active proteins including but not limited to mitogen-activated protein kinase, cyclooxygenase-2, and matrix metalloproteinases (MMPs) 1 and 3 are activated (17,18,23,24,25). MMPs are destructive to the basement membrane and potentially stimulate further destructive signals. Collectively, all of these processes lead to the fourth clinically relevant ulcerative phase that can be observed macroscopically. This culminates in the loss of epithelial tissue and ultimate breach of the mucosal barrier. Patients experience significant pain and there can also be concomitant microbial colonization, leading to increased risk of sepsis. Cell-wall products from colonizing bacteria are likely to penetrate into the submucosa, where they activate infiltrating mononuclear cells to produce and release additional proinflammatory cytokines, further amplifying inflammation (25). In the final phase, healing occurs once the insult has been removed, mediated by signaling from the extracellular matrix and mesenchyme. Interestingly, the time frame varies between individuals (17,18).
are important in mucositis development. Chemotherapy treatment has shown to increase the transcription factor AP1 (21), and this family of transcription factors is known to induce damage in all cells within the mucosa and submucosa (18). In addition, increased expression of IL-1β and TNF has been detected in the submucosal cells of hamsters following acute targeted radiation (22). In the same experiment, interference with TNF-α reduced mucositis, indicating that the cytokine signaling that is set up relatively early on plays a key role in development of injury. In the third phase of signal amplification, the proinflammatory cytokines that were upregulated earlier begin to amplify the original signal and positively feed back on NFκB, thus causing further amplification. In addition, biologically active proteins including but not limited to mitogen-activated protein kinase, cyclooxygenase-2, and matrix metalloproteinases (MMPs) 1 and 3 are activated (17,18,23,24,25). MMPs are destructive to the basement membrane and potentially stimulate further destructive signals. Collectively, all of these processes lead to the fourth clinically relevant ulcerative phase that can be observed macroscopically. This culminates in the loss of epithelial tissue and ultimate breach of the mucosal barrier. Patients experience significant pain and there can also be concomitant microbial colonization, leading to increased risk of sepsis. Cell-wall products from colonizing bacteria are likely to penetrate into the submucosa, where they activate infiltrating mononuclear cells to produce and release additional proinflammatory cytokines, further amplifying inflammation (25). In the final phase, healing occurs once the insult has been removed, mediated by signaling from the extracellular matrix and mesenchyme. Interestingly, the time frame varies between individuals (17,18).
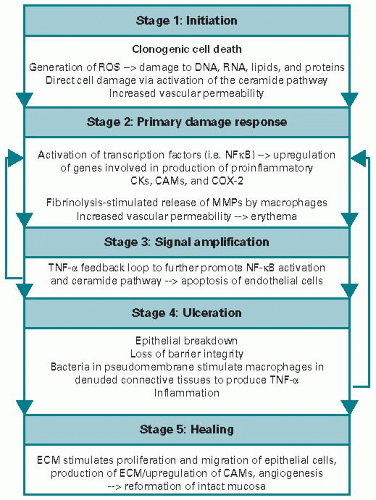 Figure 18.1. Overlapping five-phase model of AT mucositis. ROS, reactive oxygen species; NFκB, nuclear factor kappa B; CKs, cytokines; CAMs, cell adhesion molecules; COX-2, cyclooxygenase-2; MMPs, matrix metalloproteinases; TNF-α, tumor necrosis factor alpha; ECM, extracellular matrix. |
Importantly, with the development of this updated fivephase pathophysiology model, it was recognized that these “phases” occur not only in the oral mucosa but also along the entire alimentary canal. Keefe (26) proposed that these mechanisms could, and indeed should, be similar to any region of the AT, as it has the same embryological route of development involving all three germ layers of the developing embryo. The specialized differences in local function along the tract offer an explanation for why different regions are more susceptible to “early” mucositis and others are more susceptible to “late” mucositis (1,26). However, the true extent of the complexity of alimentary mucositis is still unknown, with the timing and sequence of injury events just one of a number of key issues that are not fully understood. The key underlying challenge to understanding mechanisms is the relative difficulty and invasiveness in obtaining samples from sites within the AT. Therefore, in order to obtain longitudinal data from multiple sites, animal models are necessary.
ANIMAL MODELS OF AT MUCOSITIS
Animal models of AT mucositis have provided extensive information on mechanisms. The development of appropriate animal models allows scientists to ask highly specific questions about any region of the AT. Moreover, responses to questions can be measured by only changing one variable at a time. Another key advantage of animal models is the highly homogeneous population, which reduces variability between animals, ensuring an accurate reflection of the damage that is occurring within the AT. Over the last several decades, there has been an increase in both the variety and complexity of these animal models, from basic survival endpoints to examining specialized biomarkers of damage. There have been too many experiments to name them all and their authors; however, a brief summary of the history of animal models of AT mucositis is given in Figure 18.2. It is important to recognize that animal models are highly dynamic, moving between injury-inducing agents and combinations of therapies over time. Below we have described several of the most frequently published models.
ORAL MUCOSITIS ANIMAL MODELS
Wolfgang Dorr and colleagues (27,28,29,30,31) developed a radiation mouse model in the early 1990s (Figure 18.2), which involves irradiating the tongue and snout. This animal
model results in mucosal ulceration within the treatment field of the lower tongue surface, corresponding to confluent mucositis or grade 3 of the classification of the Radiation Therapy Oncology Group/European Organization for Research and Treatment of Cancer. The mouse radiation model accurately assesses mucosal response to treatment and has been used to test a variety of agents, including keratinocyte growth factor (palifermin) (29,30,32,33,34), sodium selenite (35), and amifostine (36). In addition, it has been used to investigate the effects of combining targeted therapy, with an epidermal growth factor (EGF) receptor tyrosine kinase inhibitor (TKI), and radiation on the mucosal response (37), which is extremely important in the new era of combination therapies. The evolution of this model has also seen it being used extensively to investigate the effects of combined chemotherapy and radiotherapy (32,38). In addition, it has enabled detailed studies to be conducted comparing effects of single-dose and fractionated radiotherapy on the head (34,39,40,41,42).
model results in mucosal ulceration within the treatment field of the lower tongue surface, corresponding to confluent mucositis or grade 3 of the classification of the Radiation Therapy Oncology Group/European Organization for Research and Treatment of Cancer. The mouse radiation model accurately assesses mucosal response to treatment and has been used to test a variety of agents, including keratinocyte growth factor (palifermin) (29,30,32,33,34), sodium selenite (35), and amifostine (36). In addition, it has been used to investigate the effects of combining targeted therapy, with an epidermal growth factor (EGF) receptor tyrosine kinase inhibitor (TKI), and radiation on the mucosal response (37), which is extremely important in the new era of combination therapies. The evolution of this model has also seen it being used extensively to investigate the effects of combined chemotherapy and radiotherapy (32,38). In addition, it has enabled detailed studies to be conducted comparing effects of single-dose and fractionated radiotherapy on the head (34,39,40,41,42).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


