Fig. 10.1
Origin of MDSCs. In the bone marrow, hematopoietic stem cells (HSC) differentiate into common myeloid progenitor (CMP) followed by granulocyte-macrophage progenitor (GMP), which give rise to immature myeloid cells mainly comprised of precursors of neutrophils and monocytes. In normal condition, immature myeloid cells differentiate into macrophages, dendritic cells, and neutrophils (black arrows). However, during cancer progression the tumor microenvironment produces growth factors, cytokines, and chemokines that altered the myelopoiesis in the bone marrow. These tumor-derived factors block the differentiation of immature myeloid cell to fully mature cells creating an accumulation of pathological activated immature monocytes and neutrophils known as M-MDSCs and PMN-MDSCs, respectively (red arrows). Besides the expansion of MDSCs during tumor progressions, these cells acquire potent immunosuppressive activity. Within the tumor microenvironment, M-MDSCs differentiate to TAMs with similar ability to suppress antitumor immune responses
In humans, MDSCs have been identified and studied primarily in the peripheral blood mononuclear cell (PBMC) fraction. However, humans do not have Gr-1 antigens, which make direct comparison between mice and human MDSCs difficult. PMN-MDSCs are defined as CD11b+CD14−CD15+ or CD11b+CD14−CD66b+, whereas M-MDSCs are defined as CD11b+CD14+HLA-DR−/loCD15− [10]. There is a small population that includes a mixed group of MDSCs characterized as Lin− (including CD3, CD14, CD15, CD19, and CD56) HLA-DR−CD33+ cells. This population that comprised a more immature phenotype has been defined as “early-stage MDSCs (e-MDSCs). At the moment, the minimum requirement to define and characterize human MDSCs, including PMN-MDSCs, M-MDSCs, and e-MDSCs, should follow the phenotypic criteria [10]. Using these criteria, M-MDSCs are separated from monocytes based on the expression of HLA-DR (MHC class II molecule). On the other hand, the only method to separate PMN-MDSCs from normal neutrophils is by gradient centrifugation using a standard Ficoll gradient. PMN-MDSCs are found enriched in the low-density fraction (PBMCs), whereas neutrophils are found within the high-density fraction [11, 12]. Recently, lectin-type oxidized LDL receptor 1 (LOX-1) has been identified as a potential marker of PMN-MDSCs in humans [12]. LOX-1 expression on neutrophils could be used for direct identification of PMN-MDSCs found in blood and tumor tissue. Furthermore, LOX-1+ neutrophils isolated from the blood of cancer patients were shown to suppress T cell proliferation, whereas LOX-1− neutrophils were not suppressive.
10.3 Mechanism of MDSC-Mediated Immune Suppression
To fully identify myeloid cells as MDSCs, their functional activity needs to be tested; this is achieved usually by an in vitro suppression assay, which measures their ability to suppress the function of immune cells in tumor-bearing hosts. Activated MDSCs are implicated in the direct suppression of NK and B cells along with their primary target, T cells [13–15]. Immune suppression by MDSCs involves several mechanisms such as the increase of arginase 1 (Arg1) and inducible nitric oxide synthase (iNOS) production, the increased production of reactive oxygen and nitrogen species (ROS and peroxynitrite, PNT), and other immune-suppressive factors. Upregulation of Arg1 in MDSCs leads to the depletion and conversion of l-arginine, an essential amino acid needed for T cell proliferation, to urea and l-ornithine [16]. Under limiting amounts of l-arginine, upregulation of iNOS in MDSCs leads to the production of nitric oxide (NO) which reacts with superoxide and generates PNT [17]. Production of PNT by MDSCs causes the nitration and nitrosylation of the T cell receptor (TCR), thus disrupting potential CD8+ T cell-antigen interactions leading to T cell tolerance [18]. Also, PNT reduces the binding of antigenic peptides to MHC molecules on tumor cells and blocks T cell migration by nitrating T cell-specific chemokines, such as CCL2 [19, 20]. Another mechanism for suppression of T cell responses is the production of ROS by MDSCs. It has been shown that tumor-derived factors generate MDSCs that produced high levels of ROS which contribute to the suppressive activity of these cells [21]. Although these are the major mechanisms of immunosuppression by MDSCs, there are several other factors involved including transforming growth factor-β (TGF-β), interleukin-10 (IL-10), cyclooxygenase-2 (COX-2), indoleamine 2,3-dioxygenase (IDO), and many others. The main immunosuppressive cytokines produced by MDSCs are TGF-β and IL-10. Although TGF-β has shown to be produced by tumor cells, the production of this cytokine by MDSCs inhibits cytotoxic T cell responses in tumor-bearing mice [22]. MDSCs produced high amounts of IL-10 which impairs antitumor responses by inhibiting T cell activation [23]. In addition, MDSCs expressed several enzymes that are involved in the depletion of essential nutrition factors for T cell function. Upon stimulation by pro-inflammatory molecules, COX-2 is activated and induces Arg1 activity in MDSCs [16]. Other studies had implicated IDO expression in MDSCs which inhibit T cell function by depleting l-tryptophan and inducing T cell apoptosis within the tumor microenvironment [24]. MDSCs also block T cell function by depleting cysteine and impairing T cell activation in tumor [25]. In addition, MDSCs have the ability to induce the expansion of regulatory T cells (Treg) [26, 27]. Most of the mechanisms of suppression found to be implicated in MDSC function do not act simultaneously and are dependent on type of MDSC, type of tumors, and location of the cells.
Several recent studies have provided evidence that the ratio of PMN-MDSCs to M-MDSCs is really important because these cells utilize different mechanisms to suppress T cell responses [28]. For instance, M-MDSCs have the ability to suppress T cell activation in an antigen-specific and nonspecific manner. This suppression of T cell responses by M-MDSCs is associated with the increased expression of iNOS and production of NO [28, 29]. On the other hand, PMN-MDSCs are capable of suppressing immune responses primarily in an antigen-specific manner, which induces CD8+ T cell tolerance in the host. Immune suppression by PMN-MDSCs is associated with the increased expression of Arg1 along with high levels of ROS and PNT [8, 18]. Although PMN-MDSCs are more abundant than M-MDSCs in a tumor-bearing host, they are less immunosuppressive than M-MDSCs when compared on a per cell basis [30]. Location was also determined to be important in dictating the strength of suppression by MDSCs. Recent years have provided ample evidence indicating that MDSCs in the tumor microenvironment are more suppressive than MDSCs in peripheral lymphoid organs and peripheral blood [31, 32]. There is clear evidence now suggesting that the increased suppressive activity of MDSCs is regulated by the low levels of oxygen (hypoxia) found in tumor tissues [33]. However, more studies are needed to better understand what other tumor-associated factors and mechanisms are implicated in the potent suppressive activity by MDSCs within the tumor microenvironment (Fig. 10.2).
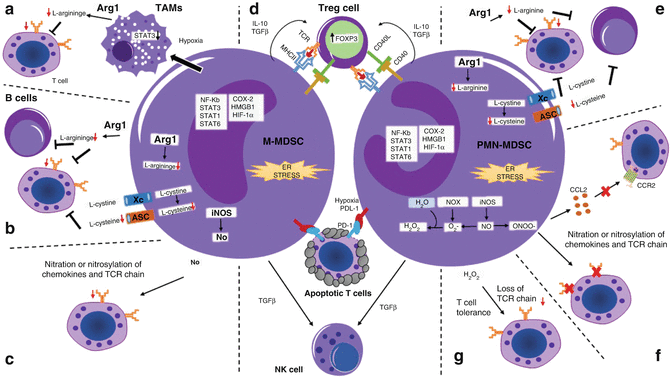
Fig. 10.2
Mechanisms of immunosuppression by MDSCs in cancer. In cancer, both M-MDSCs and PMN-MDSCs can suppress antitumor T cell responses through different mechanisms. During tumor progression, these mechanisms are regulated by several transcription factors and enzymes and by the activation of the ER stress response. (a) In hypoxia conditions, upregulation of HIF-1α induces the differentiation of M-MDSCs into TAMs in a STAT3-dependent manner. In the tumor microenvironment, TAMs suppress T cell responses by sequestering arginine through activation of Arg1. (b) M-MDSCs deprive T and B cells of essential amino acids including arginine and cysteine. (c) The main mechanism of immunosuppression by M-MDSCs is the expression of iNOS and release of NO. The release of NO causes the nitration or nitrosylation of T cell receptors as wells as chemokines that attract T cells. (d) Both M-MDSCs and PMN-MDSCs release IL-10 and TGFβ which induces the development and expansion of antigen-specific Tregs that may require CD40-CD40L interactions. Furthermore, the release of TGFβ is implicated in the suppression of NK cells by MDSCs. Within the tumor microenvironment, MDSC subsets upregulate PDL-1 expression which interact with PD-1 on T cells resulting in cell death. (e) Similarly to M-MDSCs, PMN-MDSCs suppress T cell responses by depleting arginine and cysteine from the tumor microenvironment. (f, g) PMN-MDSCs are characterized by their release of reactive nitrogen or oxygen species. (f) The production of PNT by PMN-MDSCs causes the nitration or nitrosylation of the TCR chain inhibiting T cell proliferation. Another important function of peroxynitrite release by MDSCs is the modification of CCL2, chemoattractant, which affects T cell migration. (g) Also, activation of NOX is responsible for the increased production of reactive oxygen species by tumor-associated PMN-MDSCs
10.4 Mechanisms Regulating MDSC Accumulation and Function
One of the main questions in the MDSC field is how is their expansion, accumulation, and activation regulated? Several years ago we proposed a two-signal model describing that MDSC accumulation requires two distinct types of signals [34]. This model is divided into two phases: (1) the expansion phase associated with the inhibition of their terminal differentiation and (2) the activation phase that is responsible in the conversion of immature myeloid cells into immunosuppressive MDSCs. However, we assert that these two phases partially overlap but are governed by different sets of transcription factors and intermediates (Fig. 10.3).
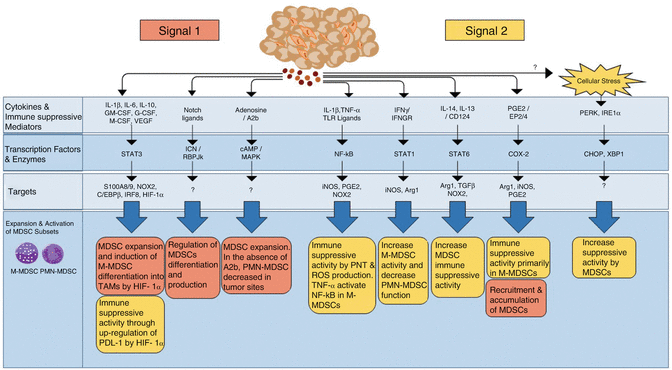
Fig. 10.3
Tumor-associated factors and molecular pathways regulating MDSC expansion and function in cancer. The tumor microenvironment produced a large number of cytokines and immune-suppressive mediators that regulate MDSC expansion and immune-suppressive activity, signals 1 and 2, respectively. Exposure of MDSCs to tumor-secreted growth factors (signal 1) and other mediators regulates several signaling pathways involved, especially STAT3, in M-MDSC or PMN-MDSC expansion and accumulation. However, for the acquisition of immune-suppressive activity by immature myeloid cells, signal 2 is required giving rise to both M-MDSCs and PMN-MDSCs. These signaling pathways are important for both PMN-MDSCs and M-MDSCs (NF-κB, STAT3, STAT6, ER stress response) or may have opposite roles in MDSC subsets (STAT1). COX-2 has been described to be more specific to M-MDSCs
The first phase is mostly driven by tumor-derived growth factors along with STAT3, IRF8, C/EBPβ, RB1, notch, adenosine receptor A2b, and NLRP3.
STAT3
Activation of signal transducer and transcription activator 3 (STAT3) in MDSCs requires myeloid-specific growth factors such as GM-CSF, G-CSF, M-CSD, IL-6, VEGF, and several other factors [35]. STAT3 plays a major role in the regulation of MDSCs. For example, tumor-bearing mice treated with different STAT3 inhibitors have shown a decrease in MDSC accumulation [36]. Interestingly, STAT3 is not only involved in the expansion of MDSCs but also in their function and differentiation; for instance, M-MDSCs are able to differentiate into tumor-associated macrophages (TAMs) within the tumor microenvironment through STAT3 regulation [37, 38].
IRF8
IFN regulatory factor 8 (IRF8) has been described as a negative regulator of MDSC differentiation. In the absence of IRF8, there is an increase in MDSC accumulation in both spleen and tumor tissues; however, overexpression of IRF8 resulted in a decreased accumulation of MDSCs in a spontaneous mouse tumor model [39, 40].
C/EBPβ
CAAT-enhancer-binding protein beta (C/EBPβ) is the only member of its family that has been implicated in MDSC expansion. Mice lacking C/EBPβ specifically in hematopoietic cells had lower frequencies of MDSCs—especially M-MDSCs—suggesting its role in MDSC differentiation [41].
Rb1
The retinoblastoma protein 1 (Rb1) has been implicated in MDSCs found in both mice and humans [42]. In tumor-bearing mice, Rb1 was shown to regulate the differentiation of M-MDSCs to PMN-MDSCs. M-MDSCs with high levels of Rb1 mainly give rise to macrophages and DCs, whereas the vast majority of M-MDSCs with low levels of Rb1 differentiate toward PMN-MDSCs. Recently, the accumulation of Rb1lo Ly6G+ PMN-MDSC was confirmed in a transgenic model of breast cancer in mice [43].
Notch Pathway
Another pathway involved in the accumulation and differentiation of MDSCs is notch signaling pathway. It has been shown that notch is downregulated upon activation of casein kinase 2 (CKII) in MDSCs. Therefore, tumor-bearing mice treated with CKII inhibitor showed an improvement in notch signaling and DC differentiation [44] These results suggests that the downregulation of notch signaling skewed the differentiation of the hematopoietic progenitors toward MDSCs instead of DCs. Recently, it has been shown that the inhibition of notch signaling enhances the generation of PMN-MDSCs but decreases the production of M-MDSCs [45].
Adenosine Receptor A2B
In the tumor microenvironment, there is an increased level of extracellular adenosine that is associated with MDSC accumulation via engagement of the adenosine receptor A2b expressed on MDSCs [46]. Tumor-bearing mice lacking A2b had lower number of MDSCs compared to wild type, suggesting that the A2b receptor is critically involved in the expansion of MDSCs, especially PMN-MDSCs [47]. This correlates with the most recent study using a melanoma model treated with an agonist and an antagonist of A2b, in which treatment with the agonist increased both tumor growth and MDSC accumulation whereas treatment with the antagonist resulted in a decreased of tumor progression with a reduction of MDSC numbers within tumor tissues [48].
NLRP3
NLR family, pyrin domain containing 3 (NLRP3) is an intracellular sensor associated with the inflammasome, which, upon activation, induces the generation of interleukin (IL)-1β and IL-18. Tumor-associated MDSCs express NLRP3, and Nlrp3−/− mice had lower levels of MDSCs within tumor tissues compared to wild-type mice, suggesting that NLRP3 may play a role in the expansion and accumulation of MDSCs [49]. However, more studies are needed to determine the exact role of NLRP3 in MDSC function. Clearly, these signaling pathways regulate MDSC expansion and are involved in blocking normal immature myeloid cell differentiation. However, the majority of these factors are implicated in the first phase of MDSC regulation but are not sufficient to promote their activation into immunosuppressive cells (Fig. 10.3).
As part of our two-signal model, the acquisition of the suppressive activity found in MDSCs is mediated by factors mostly produced by the tumor stroma including pro-inflammatory molecules such as IFNγ, IL-1β, IL-13, toll-like receptor (TLR) ligands, and others. Each of these molecules is involved in the activation of several signaling pathways associated with many factors such as NF-κB, STAT1, STAT6, prostaglandin E2 (PGE2) and cyclooxygenase (COX-2), high-mobility group box 1 (HMGB1), and hypoxia-inducible factor-1α (HIF-1α), all of which are implicated in the suppressive activity of MDSCs. Recently, the endoplasmic reticulum (ER) stress pathway has been linked to the suppressive activity of MDSCs found in cancer.
NF-κB
In MDSCs, the activation of the NF-κB pathway is predominately mediated by the TLR ligands: IL-1β or TNF-α—both of which have been shown to increase suppressive activity [34]. The role of TLR ligands in MDSC function is still not clear. Some studies have shown that TLR ligands drive the suppressive activity of MDSCs, whereas others suggest that these ligands inhibit their function [50–52]. During inflammation and cancer, IL-1β produced by tumors cells leads to the activation of MDSCs through the NF-κB pathway resulting in the increased production of PNT [14, 19, 53, 54]. Another well-known activator of the NF-κB pathway is tumor necrosis factor-α (TNF-α), which is implicated in the maturation and function of MDSCs [55]. It was shown that the transmembrane ™ form of TNF-α could increase the suppressive activity of MDSCs via its receptor and regulate iNOS expression in a NF-κB-dependent manner [56]. One study using an inflammatory mouse model showed that the role of TNF-α is restricted to M-MDSCs [57]. Also, activation of NF-κB through TNF-α receptor could promote survival of MDSCs [58]. At this moment, NF-κB can be involved in MDSC expansion, but its main role is to signal activation of these cells leading to acquisition of a suppressive phenotype, especially in M-MDSCs.
STAT1
Several studies had shown that activation of STAT1 is linked to the suppressive activity of MDSCs [17, 29, 59]. STAT1 is activated upon IFNγ stimulation and is involved in the upregulation of iNOS and Arg1 expression. Sinha et al. demonstrated that activation of STAT1 through IFNγ did not regulate MDSC accumulation or function [60]. Subsequent studies focused on the role of the IFNγ-STAT1 pathway in the suppressive activity of MDSC subsets. A recent study showed that STAT1 plays a major role by increasing the suppressive function of M-MDSCs [61]. However, activation of STAT1 through IFNγ receptors was shown to decrease PMN-MDSC function and survival [62]. These studies suggest that the IFNγ-STAT1 signaling pathway may have opposite roles in M-MDSCs and PMN-MDSCs. Indeed, this could explain why the initial study did not observe the involvement of IFNγ and/or STAT1 in the suppressive activity of MDSCs since the study of Sinha et al. analyzed the function of total MDSCs (CD11b+Gr-1+) rather than the subsets separately. It is possible that high PMN-MDSC numbers, and not M-MDSCs, compensate with the suppressive activity observed in the absence of the STAT1 signaling pathway.
STAT6
Another transcription factor involved in the function of MDSCs is STAT6. In MDSCS, the STAT6 signaling pathway is activated upon stimulation with IL-4 and IL-13 through its receptor (CD124, IL-4Rα) leading to the upregulation of Arg1 expression and production of TGF-β, a potent inhibitor of T cell proliferation [22, 63, 64]. In addition, STAT6 has been shown to be involved in the survival and accumulation of MDSCs [65].
PGE2
Prostaglandins, especially PGE2, play a major role in the suppressive activity of MDSCs in cancer. Rodriguez and colleagues found that the signaling through the PGE2 receptor E-prostanoid (EP) 4, which is expressed in MDSCs, induces Arg1 expression and its activity [16]. Furthermore, expression of the main regulator of PGE2 production, COX-2, was directly correlated with the induction of Arg1 and iNOS in tumor-infiltrating MDSCs [66]. These results were confirmed by Obermajer et al. in which they described a positive feedback loop between PGE2 and COX-2 that led to the conversion of monocytes into M-MDSCs [67]. Based on this, ex vivo MDSCs were generated using PGE2 which induce the production of suppressive factors [68]. In mouse models of mesothelioma and glioma, treatment with a COX-2 inhibitor blocked the accumulation and function of MDSCs [69, 70]. Moreover, the involvement of PGE2 in MDSC function was confirmed in melanoma patients that were treated with a COX-2 inhibitor and had MDSC-like cells with less suppressive activity [71]. Primarily, PGE2 has shown a role in the regulation of immune suppression by MDSCs, but has been implicated also in their recruitment and accumulation [72].
HGMB1
HGMB1 is a DNA-binding protein that present at high levels within the tumor microenvironment [73]. HGMB1 affects MDSC via binding to TLR4 and receptor for advanced glycation endproducts (RAGE). Signaling through both receptors activates the NF-κB pathway. Parker et al. demonstrated that HGMB1 drives the accumulation and suppressive activity of MDSCs and also promotes the survival of MDSCs by inducing autophagy in cells within the tumor microenvironment [74]. Recently, Su et al. demonstrated that in vivo HGMB1 blockade with a monoclonal antibody against HGMB1 B box decreased the accumulation of M-MDSCs in both spleen and tumor tissues [75]. These studies demonstrate that HGMB1 may play a key role in the accumulation and function of MDSCs.
HIF1α
The absence of oxygen (hypoxia) is one of the biggest differences between the tumor microenvironment and peripheral lymphoid organs. It has been shown that within the tumor microenvironment and in response to hypoxia, HIF-1α can regulate MDSC function and the differentiation of M-MDSCs to TAMs in a STAT3-dependent manner [37, 38]. More recently, Norman et al. demonstrated that HIF-1α selectively upregulate programmed death-ligand 1 (PD-L1), an immune checkpoint ligand on MDSCs [76] (Fig. 10.3).
Recent studies have suggested that ER stress responses are involved in the suppressive behavior of MDSCs in tumor-bearing hosts. The ER stress response pathway, also known as the unfolded protein response (UPR), is responsible for preserving ER homeostasis during extrinsic or intrinsic stress [77, 78]. ER stress response is comprised of three ER-localized transmembrane protein sensors: inositol-requiring enzyme 1α (IRE1α), activating transcription factor 6α (ATF6α), and double-stranded RNA-dependent kinase (PKR)-like ER-related kinase (PERK). Activation of these ER sensors leads to the upregulation of several transcription factors including the spliced X-box protein 1 (sXBP1), CCAAT-enhancer-binding protein homologous protein (CHOP), and others. Both MDSCs isolated from tumor-bearing mice and cancer patients overexpress several markers of ER stress, including sXBP1 and CHOP, and display an enlarged ER—a hallmark of ER stress [79]. Tumor-bearing mice treated with thapsigargin, an ER stress inducer, have an increased number of MDSCs with potent suppressive activity [80]. We showed that induction of ER stress with thapsigargin converts normal human neutrophils to a suppressive PMN-MDSC phenotype in an IRE1α-/XBP1-dependent manner [12]. Besides the role of IRE1α/XBP1 in MDSC function, recent findings have implicated CHOP in the suppressive activity of tumor-associated MDSCs. For example, CHOP-deficient MDSCs lose their suppressive function and acquire an immune stimulatory phenotype [81, 82]. Although CHOP-deficient MDSCs did not have the ability to suppress T cells that were stimulated in an antigen-nonspecific manner, they retained their ability to block antigen-specific T cells [83]. At this moment, only the IRE1α/XBP1 and PERK/CHOP signaling pathways have been implicated in the function of MDSCs. However, more studies are needed to understand the specific mechanisms of ER stress responses that regulate the suppressive activity of MDSCs.
10.5 Relationship of MDSCs with Other Myeloid cells
Over the years, our understanding of myeloid cells in cancer has shed light to their important role in tumor development, progression, and metastasis. The main three groups of terminally differentiated myeloid cells are macrophages, DCs, and PMNs. It is now clear that the tumor microenvironment alters myeloid cell differentiation by arresting these cells in an immature stage with potent immunosuppressive activity. Another myeloid cell involved in immune suppression is the tumor-associated macrophage (TAM) that arises from a tumor-infiltrating monocyte or M-MDSC within the tumor microenvironment. However, the relationship of MDSCs with other myeloid cells is still not clear. The main concern is the lack of specific markers for MDSCs making it difficult to discreetly identify these cells separate from monocytes and neutrophils. This is why in many previous studies, MDSC-like cells with suppressive activity were called monocytes and neutrophils. Recent data has provided evidence about the specific nature of MDSCs in cancer [1]. Immunosuppressive activity is an intrinsic feature of MDSCs. For instance, MDSCs could be generated in vitro from bone marrow progenitor cells in the presence of tumor-secreted factors. Unlike MDSCs, mature neutrophils or monocytes in the presence of these tumor-associated factors cannot suppress T cell responses in vitro [2]. Several studies, involving genomic, proteomic, and transcriptomic analysis, had provided evidence that supports the differences between PMN-MDSCs and neutrophils in tumor-bearing mice [84–88]. Moreover, a recent study provided information that suggests that PMN-MDSCs are different to neutrophils from healthy donors or cancer patients based on their gene profile [12]. In humans, PMN-MDSCs can be distinguished from neutrophils by LOX-1 expression. In mice the role of LOX-1 is not clear [3]. Within the hypoxic tumor microenvironment, M-MDSCs differentiate into TAMs, and this is regulated by HIF-1α and STAT3 activation [9, 33, 37, 38]. Both myeloid cells have potent immune-suppressive function, especially within the tumor sites; however, M-MDSCs can be distinguished from TAMs by changes in monocyte-/macrophage-associated markers. The main changes observed during M-MDSC differentiation into TAMs include an increased in F4/F80 and CD115, intermediate expression of Ly6C, and low expression of IRF8 and S100A9 protein [4, 89, 90]. MDSCs possess several biochemical features that are not observed in neutrophils or monocytes including high Arg1 and iNOS expression and activity as well as high levels of ROS and PNT production. In addition, recent studies have shown that MDSCs have an increase in the ER stress response that is linked to their suppressive function.
Among the different types of myeloid cells in cancer, there is a better understanding of the relationship between M-MDSCs and TAMs. Recent data from several groups have shown that M-MDSCs are TAM precursors within the tumor microenvironment. In addition, there are two polarized phenotypes of macrophages: the classically activated (M1 macrophages) with antitumor properties and the alternatively activated (M2 macrophages) with pro-tumor properties. The latter is more related to TAMs, but it is unclear if these two phenotypes are that of the same macrophage and what the molecular mechanism is that regulates them within the tumor microenvironment [91]. In addition, it is unknown if the polarization of macrophages is dictated by M-MDSCs before TAM differentiation. On the other hand, the nature of PMN-MDSCs and their relationship with neutrophils is a subject of discussion in different studies and an ongoing debate. Although PMN-MDSCs are widely accepted in the field, the concept of neutrophil polarization in cancer raises questions of whether these cells are pathologically similar to PMN-MDSCs [92]. Similarly to macrophages, the concept of tumor-associated neutrophils (TANs) consists of neutrophils with antitumor (N1 cells) or pro-tumor (N2 cells) properties within the tumor microenvironment [93]. Studies have implicated TGF-β and type 1 interferons (IFNs) as cytokines in the regulation of TAN plasticity in cancer [93–95]. Neutrophils by definition are short-lived and terminally differentiated cells, whereas PMN-MDSCs are immature cells. For this reason, it is hard to imagine that TANs could be polarized in the tumor microenvironment. Indeed, several studies have shown that these cells have the ability to suppress T cell responses in tumor-bearing mice, which supports these cells as PMN-MDSCs [96, 97]. However, the antitumor role of TANs has been described in cancer patients. Recently, Eruslanov et al. observed that TANs from early-stage lung cancer patients were not immunosuppressive, but rather stimulate T cell responses [98]. This study provides evidence for the role of TANs during early stages of tumor initiation. Only few studies have addressed the role of MDSCs in tumor initiation and how it regulates the immune responses at early stages. For instance, Ortiz et al. showed that the exposure of mice to cigarette smoke resulted in an accumulation of non-suppressive MDSC-like cells in various organs [99]. However, when cigarette smoke was combined with a carcinogen to promote the development of lung cancer, MDSCs presented potent immunosuppressive activity. These data suggest that MDSC function is controlled by the tumor microenvironment. Our understanding of the nature of MDSCs and their relationship with other myeloid cells during tumor development would open new therapeutic opportunities.
10.6 Therapeutic Strategies to Target MDSCs in Cancer
There is ample evidence that MDSC accumulation in peripheral lymphoid organs and tumor tissues correlates with a poor prognosis and clinical outcome in cancer patients [100]. In addition, MDSCs are implicated in resistance to anticancer therapies including the receptor tyrosine kinase inhibitor sunitinib as well as several chemotherapeutic drugs for lung cancer and multiple myeloma [101–103]. Since these cells have the ability to suppress antitumor immune responses, the success of cancer therapies, including immunotherapy, would depend on the immunosuppressive effect by MDSCs. Indeed, several clinical studies had shown an association with MDSC levels and response to immune checkpoint inhibitors, ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) [104–107]. Today, the increased knowledge of the molecular mechanisms responsible for accumulation and function of MDSCs in cancer has allowed for the development of therapeutic strategies targeting this cell population. Some strategies for targeting MDSCs include (a) elimination of MDSCs, (b) blocking the accumulation of MDSCs, and (c) inactivation of MDSCs [108] (Fig. 10.4). Several of these strategies have been developed and are currently being tested in the clinic.
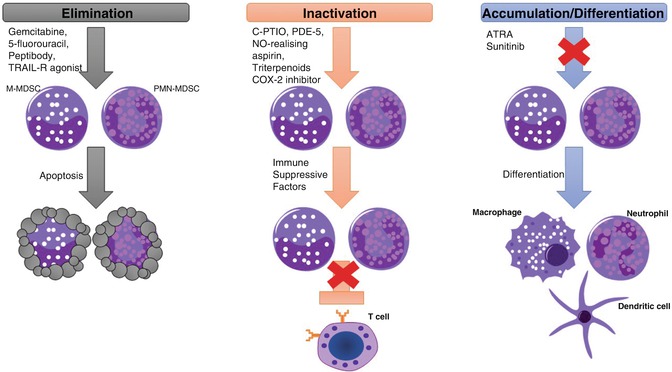
Fig. 10.4
Therapeutic strategies to target MDSCs. The main strategies to target MDSCs involved the elimination, inactivation, or blocking the accumulation and inducing the differentiation of MDSCs. First, preclinical and clinical studies have shown that MDSCs can be eliminated with low doses of chemotherapeutic drugs and with TRAIL agonist resulting in MDSC cell death. Second, in vivo inactivation of MDSCs by targeting their immunosuppressive machinery has been demonstrated in several studies. PDE5, NO-realizing aspirin, triterpenoids, and COX-2 inhibitors had shown to inhibit MDSC function by reducing ROS, RNS, and arginase levels. Finally, MDSC accumulation can be inhibited by inducing their differentiation into terminally differentiated cells with immunostimulatory activity
Elimination of MDSCs has shown to enhance antitumor efficacy of cancer immunotherapy including adoptive T cell transfer [109]. Conventional anticancer agents, in addition to their direct effect on cancer cells, have demonstrated the depletion of MDSCs in tumor-bearing hosts. Gemcitabine was the first chemotherapy agent reported to be capable of eliminating MDSCs in tumor models [110]. This study demonstrated that elimination of MDSCs by gemcitabine improves antitumor responses and enhances the effects of immune therapy resulting in tumor regression. Other chemotherapeutic drugs showing elimination of MDSCs include 5-fluorouracil, cisplatin, doxorubicin, and others [111–113]. Another strategy to eliminate MDSCs is by targeting TNF-related apoptosis-induced ligand-receptors (TRAIL-R) [79]. It was shown that upregulation of TRAIL-R, especially DR5, regulates MDSC survival in tumor-bearing host, and by using a TRAIL-R agonist, MDSCs were selectively eliminated leading to a decrease in tumor growth in a CD8+ T cell-dependent manner. Results from a phase 1 trial showed promising data that supports the use of TRAIL-R agonist in cancer patients [114]. Recently, a novel therapeutic approach to target MDSCs was developed by genetically fusing S100A9-derived peptides with the antibody Fc portion to generate a peptibody [115]. These peptibodies successfully depleted MDSCs from blood, spleen, and tumors of mouse models. However, future studies are needed to further elucidate the mechanisms of action that leads to MDSC elimination (Fig. 10.4).
MDSCs use several mechanisms to suppress antitumor immune responses, and targeting the suppressive machinery of MDSCs has been tested in cancer patients. Increased production of ROS and NO by MDSCs plays a major role in the suppression of CD8+ T cell responses. Inhibitors for the production of ROS using ROS scavengers N-acetylcysteine (NAC) and catalase are commonly used to test the function of mouse and human MDSCs ex vivo. NF erythroid 2-related factor 2 (NRF2) is a transcription factor involved in the activation of the antioxidant response and in protecting cells against damage caused by ROS. Upregulation of NRF2 by a synthetic triterpenoid was able to neutralize human MDSC activity by reducing production of ROS and dampening their suppressive function ex vivo [116]. However, a recent study from Beury et al. showed that Nrf2−/− MDSCs had a decrease in their suppressive activity compared to Nrf2+/+ MDSCs in two mouse models [117]. Scavengers of NO, such as carboxy-PTIO (C-PTIO), have been tested recently for MDSCs. Treatment with C-PTIO decreased the function of MDSCs and improved the efficacy of adoptive T cell transfer in tumor-bearing mice [118]. Besides blocking ROS and NO, another approach to inactivate MDSC function is to inhibit the catabolic enzymes upregulated by MDSCs including Arg1 and iNOS which are implicated in the suppression of T cell activation and proliferation. [63]. Inhibitors for Arg1 and iNOS have been used extensively in functional assays for MDSCs. Early studies by Rodriguez et al. demonstrated that a specific inhibitor for Arg1, N-hydroxyl-nor-l-Arg (nor-NOHA), could inhibit tumor growth and decrease Arg1 levels in MDSCs [63, 119]. Arg1 inhibitor, as well as iNOS inhibitor l-NG-monomethyl-l-arginine (l-NMMA), had shown to reduce MDSC function and improve T cell proliferation in vitro. Moreover, both Arg1 and iNOS levels in PBMCs of cancer patients have been shown to decrease in response to phosphodiesterase-5 (PDE5). Several PDE5 inhibitors had been used in the clinic to treat cancer and other conditions showing a decrease in MDSC numbers [120, 121]. Recently, a clinical report indicated that head and neck cancer patients treated with the PDE5 inhibitor tadalafil had a reduction in MDSCs and the expression of both Arg1 and iNOS, associated with an increase of T cells [122]. A parallel study demonstrated that treatment with tadalafil decreased not only MDSCs but also Tregs in blood and tumors of patients with head and neck squamous carcinoma [123]. Also, these patients treated with PDE5 inhibitor had an increase in tumor-specific CD8+ T cells. Nitroaspirin, a NO-releasing aspirin, has been shown to induce downregulation of both Arg1 and iNOS and PNT in MDSCs from a colon carcinoma model [124]. Since nitroaspirin was poorly effective as an adjuvant for adoptive T cell transfer, Molon et al. developed AT38 ([3-(aminocarbonyl)furoxan-4-yl]methyl salicylate) to inhibit reactive nitrogen species in MDSCs [20]. In this study, AT38 was shown to inhibit Arg1, iNOS, and PNT in MDSCs as well as inhibition of CCL2 nitration leading to an increase in CD8+ T cells within the tumor microenvironment. Another major mechanism of immunosuppression by MDSCs is the production of PGE2 through activation of COX-2 [125]. Several COX-2 inhibitors, including celecoxib, have demonstrated a reduction in PGE2 production and inhibition of Arg1 and iNOS in MDSCs from different mouse models [16, 17, 125]. Selective COX-2 inhibition by celecoxib reduces MDSC numbers and production of ROS and NO and improved a dendritic cell-based immunotherapy in a mesothelioma model [69] (Fig. 10.4).
Related posts:
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 Synergy Between Radiotherapy and Immunotherapy
Synergy Between Radiotherapy and Immunotherapy
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


