Chronic lymphocytic leukemia (CLL) is characterized by the accumulation of mature monoclonal B cells in peripheral blood, bone marrow, spleen, and lymph nodes. The trafficking, survival, and proliferation of CLL cells is tightly regulated by the surrounding tissue microenvironment and is mediated by antigenic stimulation, close interaction with various accessory cells and exposure to different cytokines, chemokines, and extracellular matrix components. In the last decade there have been major advances in the understanding of the reciprocal interactions between CLL cells and the various microenvironmental compartments. This article discusses the role of the microenvironment in the context of efforts to develop novel therapeutics that target the biology of CLL.
Key points
- •
CLL is characterized by the accumulation of mature monoclonal B cells in peripheral blood, bone marrow, spleen, and lymph nodes.
- •
Signals from the B-cell receptor (BCR) and the tissue microenvironment converge on several key intracellular signaling pathways, including the phosphatidylinositide 3-kinase/protein kinase B, mitogen-activated protein kinase/extracellular signal-regulated kinase, and nuclear factor-kappa B pathways, and promote leukemic cell proliferation, survival, and resistance to chemotherapy.
- •
Tissue sites provide a supportive microenvironment composed of T cells, stromal cells, cytokines, chemokines, and extracellular matrix components.
- •
The lymph node is a pivotal site of CLL cell activation and proliferation through antigenic stimulation.
- •
The dependence of CLL cells on signals from the BCR and tissue microenvironment presents opportunities for targeted therapy. Inhibitors of BCR signaling and therapeutic approaches to chemically dissect CLL cells from the supportive microenvironment have shown encouraging clinical results.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common leukemia in the Western world. An often indolent lymphoproliferative disorder, CLL is characterized by progressive accumulation of monoclonal, small, mature-appearing CD5 + B cells in peripheral blood, bone marrow, and secondary lymphoid organs. With the notable exception of allogeneic stem cell transplantation, CLL is currently an incurable disease, despite good initial responses to chemoimmunotherapy, which prolong overall survival. Current treatment modalities seem to eradicate malignant CLL cells less efficiently in bone marrow and lymph nodes than in peripheral blood. Thus, patients who initially achieve remission eventually develop recurrent disease. Moreover, the differential response of the disease in different anatomic locations indicates a significant role of the tissue microenvironment in supporting CLL cell survival, enabling them to evade the toxic effects of chemotherapy.
Recent work has demonstrated that the trafficking, survival, and proliferation of CLL cells is tightly regulated by the surrounding tissue microenvironment ( Fig. 1 ). This conclusion is bolstered by several lines of evidence. When cultured in vitro, CLL cells rapidly undergo apoptosis, but they can be temporarily rescued from programmed cell death by contact with stromal cells. Stromal cells are also known to confer a protective effect against chemotherapy-induced apoptosis. In addition, there are differences in the characteristics of CLL cells in the various tissue compartments. CLL cells in the peripheral blood are arrested in the G0/G1 phase of the cell cycle and display features that are consistent with a defect in programmed cell death and prolonged survival in vivo. Previously, this observation was thought to suggest that CLL is a malignancy of quiescent nonproliferating cells. However, recent data on telomere length and in vivo measurement of CLL cell kinetics demonstrated that CLL cells exhibit a more prominent turnover than previously appreciated. Patients with a higher proliferation rate are more likely to have active disease and clinical progression. This apparent discrepancy is because proliferation of CLL cells takes place primarily in the secondary lymphoid tissues, although it also occurs to a lesser extent in bone marrow, in areas with a vaguely nodular architecture termed pseudofollicles or proliferation centers.
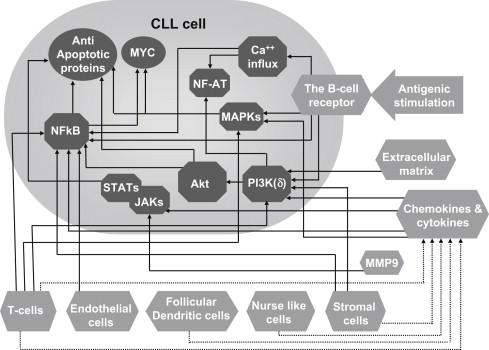
 ) including cells (eg, T cells, nurselike cells), the extracellular matrix, and enzymes (eg, MMP9) stimulate CLL cells either directly ( arrows ) or via mediators such as cytokines and chemokines ( dashed arrows ). These extracellular triggers converge into an array of intracellular biochemical responses (
) including cells (eg, T cells, nurselike cells), the extracellular matrix, and enzymes (eg, MMP9) stimulate CLL cells either directly ( arrows ) or via mediators such as cytokines and chemokines ( dashed arrows ). These extracellular triggers converge into an array of intracellular biochemical responses (  ), resulting in the upregulation of MYC and antiapoptotic proteins (
), resulting in the upregulation of MYC and antiapoptotic proteins (  ), as well as additional cellular responses. JAK, Janus kinase; MAPK, mitogen-activated protein kinase; MMP-9, metalloprotinse-9; NF-AT, nuclear factor of activated T-cells; NF-κB, nuclear factor-kappa B; PI3K, phosphatidylinositide 3-kinase; STAT, signal transducer and activator of transcription.
), as well as additional cellular responses. JAK, Janus kinase; MAPK, mitogen-activated protein kinase; MMP-9, metalloprotinse-9; NF-AT, nuclear factor of activated T-cells; NF-κB, nuclear factor-kappa B; PI3K, phosphatidylinositide 3-kinase; STAT, signal transducer and activator of transcription. The significant differences in the properties of the cells in peripheral blood and lymphoid tissues are, at least in part, explained by antigenic stimulation and close interaction with various accessory cells as well as by exposure to different cytokines, chemokines, and extracellular matrix components (see Fig. 1 ). In the last decade there have been major advances in the understanding of the reciprocal interactions between CLL cells and the various microenvironmental compartments. This article discusses the role of the microenvironment in the context of efforts to develop novel therapeutics that target the biology of CLL.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common leukemia in the Western world. An often indolent lymphoproliferative disorder, CLL is characterized by progressive accumulation of monoclonal, small, mature-appearing CD5 + B cells in peripheral blood, bone marrow, and secondary lymphoid organs. With the notable exception of allogeneic stem cell transplantation, CLL is currently an incurable disease, despite good initial responses to chemoimmunotherapy, which prolong overall survival. Current treatment modalities seem to eradicate malignant CLL cells less efficiently in bone marrow and lymph nodes than in peripheral blood. Thus, patients who initially achieve remission eventually develop recurrent disease. Moreover, the differential response of the disease in different anatomic locations indicates a significant role of the tissue microenvironment in supporting CLL cell survival, enabling them to evade the toxic effects of chemotherapy.
Recent work has demonstrated that the trafficking, survival, and proliferation of CLL cells is tightly regulated by the surrounding tissue microenvironment ( Fig. 1 ). This conclusion is bolstered by several lines of evidence. When cultured in vitro, CLL cells rapidly undergo apoptosis, but they can be temporarily rescued from programmed cell death by contact with stromal cells. Stromal cells are also known to confer a protective effect against chemotherapy-induced apoptosis. In addition, there are differences in the characteristics of CLL cells in the various tissue compartments. CLL cells in the peripheral blood are arrested in the G0/G1 phase of the cell cycle and display features that are consistent with a defect in programmed cell death and prolonged survival in vivo. Previously, this observation was thought to suggest that CLL is a malignancy of quiescent nonproliferating cells. However, recent data on telomere length and in vivo measurement of CLL cell kinetics demonstrated that CLL cells exhibit a more prominent turnover than previously appreciated. Patients with a higher proliferation rate are more likely to have active disease and clinical progression. This apparent discrepancy is because proliferation of CLL cells takes place primarily in the secondary lymphoid tissues, although it also occurs to a lesser extent in bone marrow, in areas with a vaguely nodular architecture termed pseudofollicles or proliferation centers.
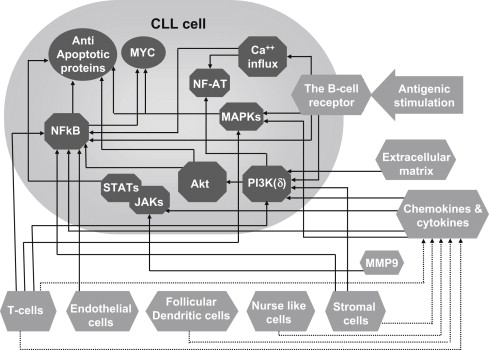
 ) including cells (eg, T cells, nurselike cells), the extracellular matrix, and enzymes (eg, MMP9) stimulate CLL cells either directly ( arrows ) or via mediators such as cytokines and chemokines ( dashed arrows ). These extracellular triggers converge into an array of intracellular biochemical responses (
) including cells (eg, T cells, nurselike cells), the extracellular matrix, and enzymes (eg, MMP9) stimulate CLL cells either directly ( arrows ) or via mediators such as cytokines and chemokines ( dashed arrows ). These extracellular triggers converge into an array of intracellular biochemical responses (  ), resulting in the upregulation of MYC and antiapoptotic proteins (
), resulting in the upregulation of MYC and antiapoptotic proteins (  ), as well as additional cellular responses. JAK, Janus kinase; MAPK, mitogen-activated protein kinase; MMP-9, metalloprotinse-9; NF-AT, nuclear factor of activated T-cells; NF-κB, nuclear factor-kappa B; PI3K, phosphatidylinositide 3-kinase; STAT, signal transducer and activator of transcription.
), as well as additional cellular responses. JAK, Janus kinase; MAPK, mitogen-activated protein kinase; MMP-9, metalloprotinse-9; NF-AT, nuclear factor of activated T-cells; NF-κB, nuclear factor-kappa B; PI3K, phosphatidylinositide 3-kinase; STAT, signal transducer and activator of transcription. The significant differences in the properties of the cells in peripheral blood and lymphoid tissues are, at least in part, explained by antigenic stimulation and close interaction with various accessory cells as well as by exposure to different cytokines, chemokines, and extracellular matrix components (see Fig. 1 ). In the last decade there have been major advances in the understanding of the reciprocal interactions between CLL cells and the various microenvironmental compartments. This article discusses the role of the microenvironment in the context of efforts to develop novel therapeutics that target the biology of CLL.
CLL cells in the context of the normal immune system
Normal B cells are programmed to rapidly respond to the environment, while causing little damage to normal tissues. They have the ability to recognize, process, and present foreign antigens to other components of the immune system, and to undergo maturation and eventually secrete antibodies directed at a specific antigen. They can undergo programmed cell death when their role is over. The reciprocal interaction of B cells with the surrounding environment leads to recruitment of cellular elements into specific tissue compartments. Furthermore, B cells migrate to various compartments that regulate their differentiation, proliferation, and survival or apoptosis. This normal immune response is achieved via multiple proteins that are produced by the B cell and the surrounding microenvironmental cells, leading to a well-orchestrated and tightly regulated sequence of events.
It is not surprising that CLL cells, the malignant counterpart of normal B cells, retain the ability to interact with their surrounding environment. However, the finely tuned orchestration and normal compartmentalization of the immune response is altered. The cause of this malignant transformation is most likely a combination of genetic predisposition and environmental triggers, leading to genetic and epigenetic changes resulting in exaggeration of positive signals and attenuation of inhibitory and pro-apoptotic mechanisms.
Interplay between tumor biology and the local microenvironment
Invasion of the primary and second lymphoid tissues by CLL cells disrupts the normal tissue architecture and physiology. The spleen and lymph nodes are diffusely infiltrated by CLL cells, whereas the bone marrow is involved in an interstitial, nodular, and/or diffuse pattern. CLL cells retain the capacity to react to a variety of external stimuli and the tissue microenvironment provides supporting signals that may differ within the various anatomic sites.
CLL cells respond to the surrounding microenvironment in vivo as demonstrated by the activation of specific signaling pathways in the tumor cells in the tissue microenvironment resulting in changes in gene expression, cellular activation, proliferation, and apoptotic threshold. In a genome wide microarray study the authors found that purified CLL cells isolated concomitantly from peripheral blood, bone marrow, and lymph nodes show characteristic gene expression profiles that reflect differential activation of signaling pathways in the various anatomic compartments. In particular, CLL cells in the lymph node upregulated more than 100 genes responsive to B-cell receptor (BCR) activation and nuclear factor-kappa B (NF-κB) signaling and are involved in proliferation. Several studies reported on comparative measurements of activation markers expressed on CLL cells and their proliferation rates in different anatomic compartments. The expression of activation markers such as CD38 and CD69 as well as proliferation are increased in CLL cells in the lymph node and bone marrow compared with circulating cells. Likewise, the antiapoptotic regulators B-cell lymphoma extra large (BCL-XL), survivin, and myeloid cell leukemia sequence 1 (MCL1) are expressed at higher levels in CLL cells in lymph nodes compared with their counterparts in peripheral blood. In addition, apoptotic priming, which describes the proximity of a cell to the apoptotic threshold, is reduced in bone marrow–resident CLL cells.
The microenvironment
Antigenic Stimulation and BCR Signaling
BCR signaling is a crucial component of normal B-cell development and plays an important role in differentiation, survival, proliferation, and antibody secretion. Conclusive experimental evidence established the importance of BCR signaling in the pathogenesis of activated B-celllike diffuse large B-cell lymphoma. Although the evidence is somewhat more circumstantial, BCR activation is now also emerging as a central stimulus in the pathogenesis of CLL. This view is based on several lines of evidence including specific BCR structures indicating a restricted antigen specificity of CLL cells, immunophenotypic characteristics shared with antigen-experienced B cells, as well as phenotypic characteristics of anergic B cells, the demonstration of ongoing BCR signaling in vivo, and a correlation of increased BCR activation or reactivity with clinical outcome.
Based on the presence or absence of somatic mutations in the immunoglobulin heavy chain variable ( IGHV ) gene expressed by clonal cells, CLL can be divided into 2 main subgroups. Expression of a mutated IGHV identifies a subtype that follows a stable or slowly progressive course, whereas expression of an unmutated IGHV gene is associated with progressive disease and inferior survival. In addition, CLL cells use a restricted repertoire of IGHV genes, which encode part of the antigen interacting domains of the BCR. Thus, preferential usage of certain IGHV genes indicates a role for antigen selection in the development of the disease. Furthermore, some cases express virtually identical BCRs, so-called stereotyped BCRs, that recognize shared antigens. These antigens remain incompletely defined but in many cases may be the target antigens of so-called polyreactive or natural antibodies, including microbial antigens and autoantigens expressed by dying cells. In the case of unmutated CLL, it is believed that the particular molecular motifs involved in tumor development are autoantigens; this view is supported by the observation that most of the CLL clones exhibiting stereotyped BCRs also demonstrate unmutated IGHV genes, as well as several studies of soluble immunoglobulins (Igs) demonstrating polyautoreactivity in unmutated CLL. In contrast, stimulation via foreign antigen is likely to underlie the pathogenesis of mutated CLL, which exhibits less structural restriction of the BCR. At present, our understanding of where B cells encounter antigen is incomplete. However, the secondary lymphoid tissues are likely to be the major anatomic site for BCR-antigen interaction. Antigens arriving through the lymph flow are sequestered and immobilized by cellular elements in the lymph nodes, thus providing an optimal setting for BCR stimulation. This is consistent with our observation of stronger BCR activation on CLL cells on the lymph nodes compared with blood or bone marrow.
BCR signaling can be broadly divided into 2 main types; one that is antigen independent or tonic and another that is antigen-mediated ( Fig. 2 ). Tonic signaling is mediated via phosphatidylinositide 3-kinase (PI3K)α and PI3Kδ, whereas antigen-dependent signaling involves activation of PI3Kδ in addition to several tyrosine kinases and adapter molecules. Antigen-dependent signaling is initiated by the tyrosine kinases LYN and spleen tyrosine kinase (SYK). In vitro studies have shown that BCR engagement on CLL cells triggers an intracellular signaling cascade leading to calcium mobilization, activation of the mitogen-activated protein kinase kinase/extracellular-signal regulated kinase (MEK/ERK), AKT/mammalian target of rapamycin (mTOR), and NF-κB pathways, and upregulation of the antiapoptotic proteins MCL1, BCL-XL, and X-linked inhibitor of apoptosis (XIAP) (see Fig. 1 ).
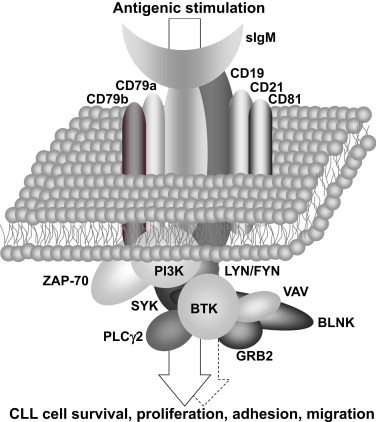
Recently, a type of BCR signaling that seems to be unique to CLL cells has been described. Specifically, Duhren-von Minden and colleagues showed that epitopes in the framework region of surface immunoglobulins (sIg) expressed on CLL cells serve as autoantigens, raising the possibility of autostimulation of the leukemic cells. Building on previous work demonstrating that the pre-BCR is able to induce ligand-independent cell-autonomous signaling by binding to an intrinsic pre-BCR glycosylation site, the group found that the BCRs of CLL cells constitutively signal even in the absence of any added antigen. This type of signaling was equally demonstrated for BCRs derived from mutated and unmutated CLLs, and seemed to be cell autonomous in that it was demonstrable on isolated individual cells. Such autonomous activation was not found with BCRs derived from multiple myeloma or other lymphoma cells. The heavy-chain complementarity-determining region (HCDR3) of the BCR was identified as the crucial interacting unit, because its insertion into a nonautonomously active BCR resulted in autonomously driven signaling. The investigators then showed that the HCDR3 interacts with an intrinsic motif in the framework region 2 (FR2) of the sIg’s VH region. Mutations in this internal FR2 epitope abrogated any autonomous signaling, demonstrating that this epitope acts as an autoantigen binding to CLL BCRs. This finding does not negate the important role of the classic model of extrinsic antigen in the pathogenesis of CLL. Further study is warranted to define the respective roles of the 2 modes of BCR signaling in CLL.
The responsiveness of CLL cells to BCR activation in vitro is heterogeneous. IGHV unmutated CLL cells are typically BCR signaling competent, whereas IGHV mutated CLL cells respond weakly or not at all to BCR crosslinking induced by anti-IgM antibodies. The Zeta-associated protein of 70-kDa (ZAP-70), a transducing signaling kinase downstream of the T-cell receptor, is expressed in most cases of IGHV unmutated CLL but less frequently in IGHV mutated CLL. Expression of ZAP-70, akin to IGHV mutation status, serves as a powerful prognostic marker and correlates with a more aggressive disease course. ZAP-70 expression is associated with increased BCR signaling in vitro. The nonresponsiveness to BCR activation in some CLL cells is reminiscent of anergized B cells and suggests that these CLL cells are chronically stimulated by antigen in vivo. Consistent with this view is the low expression of surface IgM in CLL compared with normal B cells and the recovery of BCR responsiveness after prolonged culture in vitro. Furthermore, CLL cells that do not respond to surface IgM crosslinking respond to anti-IgD or anti-CD79a antibodies, indicating that the intracellular signaling pathway is functional. Nevertheless, it has become clear that the presence of ZAP-70 enhances BCR responsiveness. This effect of ZAP-70 is independent of the kinase domain, but requires recruitment of ZAP-70 to the BCR. The demonstration of decreased internalization of an activated BCR in a B-cell line that was engineered to express ZAP-70 provides a link between decreased IgM expression that correlates with the absence of ZAP-70 and an anergic phenotype. Thus, one effect of ZAP-70 might be to interfere with anergy by maintaining higher IgM expression.
In vitro, BCR crosslinking protects CLL cells from apoptosis primarily through the PI3K/AKT pathway and increased expression of MCL1. BCR triggering also upregulates adhesion and costimulatory molecules, and increases CLL cell migration in response to the chemokines CXCL12 and CXCL13. Moreover, BCR signaling likely plays a central role in promoting CLL cell proliferation. In vitro engagement of the BCR in CLL cells induces expression of MYC, cyclin D2, and cyclin-dependent kinase 4 (CDK4). Although BCR activation promotes G1 cell cycle progression, cell division is not induced. Presumably additional costimulatory signals such as CD40L and interleukin (IL)-4, provided in the tissue microenvironment, are required.
Cell-Cell Interactions
Most studies exploring the cellular interactions in CLL have been performed using peripheral blood cells. Experimental methods relying on the investigation of circulating CLL cells in isolation lack the ability to appropriately mimic the complex cellular interactions occurring in the lymphatic niche. Despite this limitation, many in vitro observations have elucidated the in vivo crosstalk between CLL cells and nonmalignant cells. In the tissue microenvironment, CLL cells reside in close contact with T lymphocytes, stromal cells, endothelial cells, follicular dendritic cells, and macrophages. Interactions between these components regulate CLL cell trafficking, survival, and proliferation in a manner that may be partly dependent on direct physical cell-to-cell contact or mediated through the exchange of soluble factors (see Fig. 1 ).
T Cells
The interaction between CLL cells and T cells is an important component of the malignant process. First, T cells are important for CLL cell proliferation. This has been directly demonstrated in a xenograft murine model of CLL in which activated CD4 + T cells were required for CLL cell proliferation. In patients with CLL, T cells, predominantly of the CD4 + type, often make up a substantial fraction of the lymphoid infiltrate in the bone marrow and lymph nodes, where they are located both around and within proliferation centers. CD40, a key regulator of B-cell–T-cell interaction, is stimulated by CD4 + T cells expressing CD154, the ligand for CD40, that are preferentially colocalized with CLL cells in pseudofollicular proliferation centers. CLL cells activated in vitro via CD40, alone or in combination with IL-4, enter the cell cycle and are rescued from both spontaneous and drug-induced apoptosis. CD40 signaling in CLL cells induces antiapoptotic molecules such as MCL1, BCL-XL, BFL1, and survivin. Promotion of CLL cell survival and proliferation by CD40 signaling is mediated through the PI3K/AKT, MEK/ERK, and NF-κB pathways. Survivin, a member of the family of inhibitor of apoptosis proteins (IAPs), which is preferentially expressed in the large proliferating CLL cells interspersed with T cells in lymph node pseudofollicles, integrates apoptosis resistance and proliferation.
In addition to interactions mediated through direct cell-cell contact, T cells also secrete soluble factors that may contribute to CLL cell growth and survival. IL-4 inhibits spontaneous and drug-induced apoptosis in CLL cells via a mechanism involving BCL-2 upregulation. Both IL-2 and tumor necrosis factor (TNF)α variably induce CLL cell proliferation. Similarly, interferon (IFN)γ, IFNα, and IL-13 were also shown to support CLL cell survival.
Second, CLL cells can modify the cellular immune system to evade immune surveillance. Mechanisms are most likely multifactorial including production of immune-suppressing cytokines such as tumor growth factor-β and IL-10, and expression of reduced levels of adhesion and costimulatory molecules, as well as increased numbers and altered function of regulatory T cells. Gene expression profiling of purified T cells from patients with CLL revealed changes in genes involved mainly in cell differentiation, cytoskeleton and vesicle formation, trafficking, and cytotoxicity that contribute to decreased immune response. Accordingly, CD4 + and CD8 + T cells in CLL show impaired ability to form immunologic synapses, which is induced by direct cell contact with CLL cells and is mediated through tumoral expression of CD200, CD270, CD274, and CD276.
Stromal Cells
Mesenchymal stromal cells (MSC) are another important cellular component of the tissue microenvironment. Early studies exploring CLL-stromal cell interactions relied on bone marrow-derived stromal cells. These cells consist of a heterogeneous population of cells that provide structural and functional support for normal hematopoiesis. Since then, other types of human and murine MSCs have been shown to exhibit similar effects on CLL cells. Stromal cells produce and secrete various cytokines, chemokines, proangiogenic factors, and extracellular matrix components, and express surface receptors that predominantly regulate CLL cell migration and survival. The CLL-MSC crosstalk is bidirectional; thus, tumor cells are not only being supported by stromal cells but also are capable of activating and inducing stromal cell proliferation and secretion of mediators that sustain and intensify the malignant process. In the lymphoid tissues of patients with CLL, stromal cells are diffusely located throughout the tissue and in perivascular areas where they admix with CLL cells. The stromal cells are highly productive of CXCL12 and are markedly positive for α-smooth muscle actin (αSMA), a marker induced in myofibroblasts that have been activated by tumor stromal-specific growth factors. CLL cells cocultured with bone marrow stromal cells are rescued from both spontaneous and drug-induced apoptosis, in a mechanism dependent on direct cell-cell contact. Murine fibroblast cell lines have been shown to protect CLL cells from apoptosis by maintaining expression of the antiapoptotic proteins BCL-XL, XIAP and flice inhibitory protein long (FLIP L ) in the leukemic cells. Furthermore, cell-cell interactions activate the NF-κB pathway in a PI3K-dependent manner. CXCL12, secreted from stromal cells, guides CLL cell migration toward the stromal layer and promotes penetration beneath it, a phenomenon called pseudoemperipolesis.
Both cell surface receptors and extracellular matrix elements were reported to be responsible for the enhanced survival of CLL cells that are in contact with the stroma layer. Adherence of CLL cells to stromal cells is mediated simultaneously through integrins β1 and β2. MSCs highly express vascular cell adhesion molecule 1 (VCAM-1). Binding of α4β1 integrin (CD49d/CD29 or VLA-4) to either VCAM-1 or to the extracellular matrix component fibronectin rescues CLL cells from both spontaneous apoptosis and fludarabine-induced apoptosis, through PI3K/AKT signaling and BCL-X L upregulation.
Another aspect of CLL cell interaction with the stroma layer involves metalloproteinase (MMP)-9, vascular endothelial growth factor (VEGF), and endothelial cells. MMP-9, the major MMP produced by CLL cells, promotes their extravasation and lymphoid tissue infiltration through proteolytic degradation of basement membranes and extracellular matrix components. Independently of its proteolytic activity, MMP-9 also partially mediates CLL cell survival in bone marrow–derived stromal cell coculture. Binding of MMP9 to α4β1 and CD44v in CLL cells results in LYN and signal transducer and activator of transcription (STAT) 3 activation and induction of MCL1. Expression of MMP9 in CLL cells is regulated through α4β1 integrins and CXCL12. In this respect, CLL cells in the bone marrow and lymph nodes acquire and express higher levels of surface MMP-9 than could be attributed to tumor cell activation in the tissue microenvironment or derived from their adjacent accessory cells. The proangiogenic molecule VEGF also decreases spontaneous or drug-induced apoptosis of CLL cells through upregulation of MCL1, XIAP, and STAT3 signaling. In cocultures of CLL cells with bone marrow–derived stromal cells, vast amounts of VEGF seem to be secreted from the stromal cells and VEGF blockade results in decreased CLL cell survival. CLL cells cocultured with human vascular endothelial cells are also protected from apoptosis in a mechanism involving NF-κB–mediated upregulation of B-cell CLL/lymphoma 2 (BCL2), MCL1, and BCL-XL. Endothelial cells further increase expression of CD38 and CD49 in CLL cells in an NF-κB–dependent mechanism. In addition, activation of CD44 on CLL cells by extracellular matrix components such as hyaluronic acid can promote CLL cell survival through activation of the PI3K pathway.
Follicular Dendritic Cells
The literature on the role of follicular dendritic cells (FDCs) in CLL is limited. FDCs are accessory cells within normal germinal centers that retain intact antigen-antibody complexes on their cell surface and present these antigens to B cells. Normal germinal center B cells that bind to the immune complexes survive and differentiate into either memory B cells or plasma cells. FDCs are normally detected in secondary lymphoid tissue but not in bone marrow. In CLL, FDCs are seen in the lymph nodes, pseudofollicles, and in bone marrow of patients with nodular involvement. FDCs secrete several important prosurvival factors and growth factors (eg, B cell-activating factor (BAFF) and IL-15) and express other important adhesion molecules such as VCAM-1, intercellular adhesion molecule 1 (ICAM-1), plexin B1, and CD44. The effect of FDCs on CLL cells was studied using an FDC cell line (HK cells); the HK cells were shown to rescue CLL cells from spontaneous and drug-induced apoptosis in a manner that was dependent on direct cell-cell contact and associated with an increase in MCL1.
Tissue Macrophages, Monocytes, and Nurselike Cells
An intriguing example of the distinct ability of CLL cells to affect normal cellular elements, leading to the loss of normal compartmentalization and spatial control of the immune response, is the recently described phenomenon of the interaction between CLL cells and nurselike cells (NLCs). NLCs are an in vitro model believed to represent a counterpart of tissue-associated macrophages in vivo. In long-term cultures of peripheral blood mononuclear cells from patients with CLL, large, round, occasionally binucleated CD68-expressing cells grow out. NLCs were so named because CLL cells surround these cells, giving them a survival advantage. Cells with similar phenotype are also detected in vivo in secondary lymphoid tissues of patients with CLL, thus strengthening their biological relevance; yet, their numbers in the tissues are probably low. NLCs actually differentiate from monocytes and their differentiation is dependent on cell-cell contact with CLL cells. Monocytes obtained from normal donors cocultured with purified CLL cells also differentiate into NLCs but normal B cells do not induce differentiation of monocytes into NLCs. Coculturing CLL cells with NLCs protects CLL cells from spontaneous and drug-induced apoptosis in a mechanism that is partially mediated through an increase in MCL1 expression. NLCs produce and secrete chemokines and growth factors including CXCL12 and CXCL13 as well as BAFF and a proliferation-inducing ligand (APRIL), which attract CLL cells into the tissue compartment and support their survival and proliferation.
BAFF and APRIL are TNF superfamily members that are important for B-cell differentiation and survival. CLL cells themselves also express BAFF and APRIL and their receptors. However, NLCs express higher levels of BAFF and APRIL than CLL cells. BAFF binds to BAFF receptor (BAFF-R), B-cell maturation antigen (BCMA), and transmembrane activator calcium modulator and cyclophilin ligand interactor (TACI), whereas APRIL binds only to the latter 2 receptors. BAFF and/or APRIL decrease both spontaneous and drug-induced apoptosis of CLL cells. Coculturing CLL cells with NLCs in the presence of a decoy receptor that binds both BAFF and APRIL partially abolishes the protective effect of the NLCs on the viability of the CLL cells. Recently, it has been shown that BAFF, in cooperation with MYC, can lead to the development of a CLL like lymphoproliferation in mice. MYC and its target genes are upregulated in CLL cells in the lymph node and MYC is upregulated by BAFF and by BCR engagement in vitro. CLL cells that highly express MYC are more susceptible to apoptosis and can be rescued by BAFF.
Additional interactions between CLL cells and accessory cells in the tissue microenvironment involve the ligation of CD38 to CD31 and of CD100 to plexin B1. CD38 levels on the surface of CLL cells are variable and high CD38 expression is a poor prognostic factor in CLL. In proliferation centers, CD38 is upregulated in CLL cells exposed to activated T cells expressing CD40L. CD31, the ligand for CD38, is expressed on endothelial cells and NLCs. CD31 induces proliferation and prolongs the survival of CD38 + CLL cells. CD100, a transmembrane protein that belongs to the semaphorin family, is expressed on CLL cells. The high affinity receptor for CD100, plexin B1, is expressed on bone marrow stromal cells, FDCs, NLCs, and activated T cells. It has been demonstrated that engagement of CD100 by plexin B1 increases CLL cell proliferation and prolongs survival.
Trafficking and homing of CLL cells into lymphoid tissues
Chemokines consist of 2 major subgroups. One group of homeostatic chemokines is constitutively produced and secreted within the tissue microenvironment and serves to maintain physiologic trafficking. The second group includes inflammatory chemokines, which are primarily induced in inflamed tissues to recruit effector cells. Serum levels of some of the chemokines or their cognate receptors are highly increased in patients with CLL and a more effective chemotactic response is a characteristic of more aggressive subtypes of CLL cells. The responsiveness of circulating CLL cells to chemokine stimulation might facilitate the trafficking, homing, and invasion of leukemic cells into the nourishing tissue microenvironment.
An example of a chemokine that critically regulates CLL cell migration is CXCL12, a homeostatic chemokine that plays a critical role in normal trafficking and homing. CXCL12 is constitutively secreted at high levels by stromal cells and in vitro by NLCs. CXCR4 (CD184), the receptor for CXCL12, is highly expressed on circulating CLL cells, which migrate more efficiently toward CXCL12 than normal B lymphocytes. CLL cells expressing CD38 and/or ZAP-70 show stronger intracellular signaling and better chemotaxis in response to CXCL12 than cells with no CD38 or ZAP-70 expression.
CXCL13 is another homeostatic chemokine, which, along with its cognate receptor, CXCR5, plays a central role in the recruitment of B cells into the B-cell zone of secondary lymphoid organs. CXCL13 is constitutively secreted by stromal cells in the B-cell areas of the secondary lymphoid tissues. CXCL13 is expressed in vitro by NLCs as well as in vivo in the CD68 + macrophages present in CLL lymph nodes. Serum CXCL13 levels are higher in patients with CLL compared with healthy individuals and CXCR5 is also highly expressed in CLL cells.
CCL19 and CCL21 are chemokines that regulate the recruitment of lymphocytes into the T-cell zone areas of the secondary lymphoid tissues through ligation to their cognate receptor CCR7. CCL19 and CCL21 are detected in the stroma and in the high endothelial venules of lymph nodes in CLL, and the latter are an important route of lymphocyte entry into secondary lymphoid tissue. Circulating CLL cells express high levels of CCR7, which are higher in ZAP-70 + CLL cells and in patients with prominent lymphadenopathy. CLL cells that are ZAP-70 + or from patients with marked lymphadenopathy migrate more efficiently toward these chemokines.
CLL cells are not only capable of responding to cellular elements in the tissue microenvironment but also actively recruit cells from the microenvironment to their immediate vicinity. This involves CLL cell secretion of chemokines such as CCL17, CCL22, CCL3, and CCL4. CCL22 and CCL17 are T-cell attracting chemokines induced in CD40-activated CLL cells in the lymph nodes and bone marrow. Thus, CLL cells can attract CD4 + T cells to the bone marrow and lymph nodes that augment tumor cell proliferation and survival and further induce release of CCL22, creating a positive feedback loop that further promotes the malignant process.
CCL3 and CCL4 are proinflammatory chemokines crucial for the response to infection, the mediation of inflammation and the recruitment of monocytes and T cells from the blood into the tissue compartments. CLL cells secrete these 2 chemokines in response to activation of BCR and CD38 as well as during coculture with NLCs. Expression of CCL3 and CCL4 in CLL cells during coculture with NLCs positively correlates with ZAP-70 positivity. CCL3 and CCL4 are also overexpressed in CD38 + CD49d + CLL cells more than tin CD38 – /CD49d – cells. Both CCR1 and CCR5, which are the cognate receptors for CCL3 and CCL4, are expressed on monocytes and macrophages and induce their migration. Consistently, higher numbers of tumor-infiltrating CD68 + macrophages were detected in the bone marrow of patients with CD38 + CD49d + CLL. CCL3 may also indirectly protect CLL cells from apoptosis via induction of VCAM-1 in endothelial cells. Plasma levels of CCL3 and CCL4 are increased in patients with CLL compared with healthy individuals and high CCL3 levels correlate with poor prognosis in CLL.
Signaling pathways activated in the tissue microenvironment
Given that CLL cells respond in vitro to a wide variety of external stimuli (see Fig. 1 ), it is a great challenge to determine which signaling pathways are the most relevant in vivo. Clearly, engagement of the BCR, via either autonomous or extrinsic mechanisms provides a signal. The PI3K/AKT, MAPK/ERK, NF-κB, WNT, Janus kinase (JAK)/STAT, and NOTCH signaling pathways have been reported to mediate survival and/or proliferation of CLL in vitro. In particular, the PI3K/AKT, MEK/ERK, and NF-κB pathways have been shown to be activated in the tissue microenvironment. Because the composition of the microenvironment can vary between tissues, different signaling pathways may be engaged in different locations. This may be particularly the case for the lymph nodes and for the so-called proliferation centers. The activity of many intracellular signals seems to be stronger and/or more sustained in patients with more aggressive subtypes of the disease and correlates with enhanced tumor proliferation and more rapid disease progression. In particular, PI3K/AKT signaling regulates CLL cell survival and trafficking. The PI3K/AKT pathway mediates the chemotactic response of CLL cells toward CXCL12, CXCL13, CCL19, and CCL21 and promotes CLL cell survival in response to a variety of external stimuli including BCR activation, CD40L, VCAM-1, CCL19, CCL21, and many others. The inhibition of apoptosis in CLL cells through PI3K/AKT signaling is partially dependent on activation of NF-κB and upregulation of antiapoptotic genes (eg, BCL-XL and BFL-1). The classic NF-κB pathway has been shown to be activated in CLL cells in the lymph node more than in those derived from peripheral blood. A variety of in vitro stimuli induce NF-κB activity in CLL cells including engagement of the BCR or CD40, exposure to cytokines such as BAFF or APRIL, and coculture with stromal cells or endothelial cells. Compared with normal B cells, CLL cells overexpress antiapoptotic proteins such as BCL-2 and MCL1 and the resistance to apoptosis is further enhanced in the lymphoid tissues via upregulation of BCL-2 family molecules including MCL1 BCL-XL and survivin. MCL1 expression in CLL cells is commonly regulated through PI3K/AKT signaling. BCL-XL can be upregulated in CLL cells through BCR and CD40 signaling or through VCAM-1, stromal cells, and endothelial cells. The MAPK/ERK pathway also transmits prosurvival signals in CLL cells, as demonstrated in response to stimulation with CXCL12, CXCL13, CCL19, and CCL21. In addition, the MEK/ERK pathway is an important regulator of cell cycle progression and proliferation. MEK1/2 activity is important for MYC expression and S-phase entry of CLL cells. MEK/ERK mediates MYC expression in response to engagement of the BCR and Toll like receptor (TLR) 9 (induced in vitro by CpG oligonucleotide) and with BAFF stimulation. Accordingly, both phosphorylated ERK and MYC are mostly expressed in large proliferating CLL cells confined to the proliferation centers within the lymph nodes. MYC can contribute to genomic instability by selecting cells with defective DNA damage response, and its expression may be a driver of clonal evolution.
Cell proliferation is regulated by D-type cyclins that bind to CDK4 and CDK6, resulting in the phosphorylation of the retinoblastoma protein and the G1-S phase transition of the cell cycle. Cyclin D2 is overexpressed in CLL cells, especially in cells residing in the lymph nodes. IgM ligation induces cyclin D2 and CDK4 in CLL cells. The downregulation of the cell cycle inhibitor p27 and progression of CLL cells into the S phase are probably dependent on additional costimulatory signals, such as CD40 ligand and IL-4, that are provided mainly by T-helper (Th) lymphocytes in the proliferation centers of the lymph node. Accordingly, within CLL cells in the proliferation centers, cyclin D2 is highly expressed and p27 is downregulated. Cyclin D2 expression is regulated either directly through NF-κB or indirectly by MYC. Consistent with this finding is the observation that NF-κB activity is increased in the lymph nodes and particularly enhanced in CLL cells within the proliferation centers.
Models of the CLL microenvironment
Modeling tumor-host interactions is an area of intensive investigation. Such models are of particular interest given that tissue-resident CLL cells are not readily available. Currently, the most widely used in vivo model for CLL is the transgenic TCL1 mouse, in which the human TCL1 gene is expressed under the control of the Ig heavy chain variable region promoter and enhancer. TCL1 is an oncogene commonly activated in mature T-cell lymphomas that enhances AKT signaling. Onset of disease occurs late in life and the tumor cells in TCL1 transgenic mice are relatively large lymphoid cells, expressing unmutated IGHV genes. There is evidence for a role of BCR signaling in this model and a dysregulation of the T-cell compartment similar to what has been described in human CLL. The TCL1 transgenic model has also been used successfully to study novel therapeutic approaches (discussed later). In contrast to the transgenic model, New Zealand Black (NZB) mice spontaneously develop autoimmunity and B-cell hyperactivity early in life, and a CLL like disease manifests later in life. The late-onset clonal disease is of the IGVH unmutated type, which is also ZAP-70 positive. NZB mice were found to harbor a point mutation in the 3′-flanking sequence of pre–mir-16-1, which results in decreased levels of miR-16 in lymphoid tissues. This is reminiscent of the most common chromosomal lesion in human CLL: a deletion of the 13q14 chromosomal region containing the mir-15a/16-1 and DLEU2 genes. Recently, Klein and colleagues showed that deletion of the 13q14-minimal deleted region (MDR) harboring the DLEU2/miR-15a/16-1 cluster in mice results in the development of a condition that resembles human CLL. The leukemic cells of these mice express unmutated IGHV genes and some of them even present with BCRs showing stereotypical antigen-binding regions. Other transgenic mice models of CLL include (NZB × NZW)F1 mice programmed to express IL5, mice overexpressing both BCL2 and a TNF receptor-associated factor, and MYC/BAFF transgenic mice.
A complementary approach has been to xenograft the Mec-1 cell line or primary CLL cells into immune-compromised mice. Recently, Bagnara and colleagues reported that peripheral blood mononuclear cells (PBMCs) from patients with CLL xenografted into NOD/scid/γc null (NSG) mice localized and proliferated primarily in the murine spleen. These investigators found that proliferation of CLL cells in vivo was dependent on coengrafted human T cells. Furthermore, by comparing CLL cells isolated from spleens of xenografted mice to CLL cells from human blood and lymph nodes, Sun and colleagues showed that the murine spleen microenvironment supports CLL cell proliferation and activation to a similar degree as the human lymph node, which notably includes activation of BCR and NF-κB signaling in the xenografted cells. The model was then used to test the in vivo effects of ibrutinib, a Bruton tyrosine kinase (BTK) inhibitor in clinical development. Ibrutinib inhibited BCR and NF-κB signaling induced by the microenvironment, decreased proliferation, induced apoptosis, and reduced the tumor burden in vivo. Thus, these data indicate that the spleen of xenografted NSG mice can sufficiently model the role of the human microenvironment on CLL cells to make this a valid model for investigations of tumor microenvironment interactions and the evaluation of possible novel treatment approaches.
Targeting the microenvironment in CLL
The increasing appreciation of the role of the microenvironment in supporting CLL cell proliferation and survival as well as its contribution to chemoresistance has informed novel therapeutic approaches. Accordingly, a major effort is underway to find efficient ways to chemically dissect CLL cells from the microenvironmental signals, by either blocking extracellular triggers or abrogating intracellular signaling ( Fig. 3 ). In recent years, different compounds have been developed that are able to antagonize surface receptors or cytokines including small molecules that target signaling kinases and antiapoptotic proteins. Clinically, impressive responses characterized by diminished lymphadenopathy and splenomegaly have been observed with some of these novel agents.
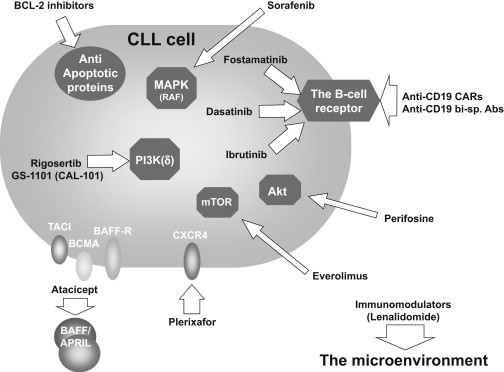
CXCR4 Receptor Antagonists
CLL cells in the peripheral blood express high levels of the surface receptor CXCR4, which signals for chemotaxis, polymerization of actin, and migration through the vascular endothelium. Blockade of the CXCR4-CXCL12 axis using CXCR4 receptor antagonists such as plerixafor (AMD3100) or T140 can efficiently antagonize CXCL12-mediated signaling and chemotaxis as well as stroma-mediated protection from both spontaneous and drug-induced apoptosis. The clinical use of plerixafor in CLL is investigated in combination with rituximab. Preliminary results showed that this agent induces dose-dependent mobilization of CLL cells to the peripheral blood.
Targeting BAFF and APRIL
As previously mentioned, BAFF and APRIL signaling through their cognate receptors BAFF-R, TACI, and BCMA are important for normal B-cell survival and may play a role in CLL. Atacicept is a recombinant soluble form of the extracellular binding domain of TACI. By acting as a molecular decoy, it neutralizes the effects of BAFF and APRIL, blocking the activation of TACI, BCMA, and the BAFF receptor. A phase Ib study of atacicept demonstrated that intravenous doses of up to 27 mg/kg were well tolerated in 21 patients with refractory or relapsed CLL with 1 partial response (PR) (overall response rate [ORR] 5%) in the highest dose cohort.
Targeting BCR Signaling
The pivotal role of BCR signaling in CLL pathogenesis points to this pathway as an ideal target for novel anti-CLL therapy (see Fig. 3 ). Small-molecule drugs targeting SYK, BTK, or PI3K isoform p110delta (PI3Kδ) show impressive results in patients with relapsed/refractory CLL as well as in treatment-naive CLL (reviewed in ). In the first few weeks of treatment with these agents, responses typically manifest with a substantial regression in lymphadenopathy that is frequently paralleled by transient lymphocytosis. The early increase in the circulating lymphocytes is assumed to reflect redistribution of the CLL cells from the lymphatic tissues into the circulation, as a consequence of disruption of mechanisms involved in migration and retention of the tumor cells in their protective tissue microenvironments. Continued treatment over the course of months results in a gradual decrease in lymphocytosis and a deepening of responses with a high rate of remission achieved with increasing duration of treatment. The clinical responses seen with these kinase inhibitors are attributed to the combined effects of direct cell cytotoxicity, inhibition of survival pathways, and impairment of CLL cell trafficking and tissue retention.
LYN Inhibitors
LYN, an SRC family nonreceptor tyrosine kinase, plays an important role in initiation as well as in the termination of BCR signaling. This dual role of LYN is due to its propagation of the BCR signal via phosphorylation of SYK and its concurrent activation of inhibitory phosphatases that terminate the response. Dasatinib, which was originally approved for treatment of chronic myeloid leukemia, is an oral kinase inhibitor primarily targeting ABL and SRC kinases but it also inhibits other kinases, including BTK. In preclinical studies, dasatinib has been shown to induce apoptosis of CLL cells that was associated with reduction in MCL1 and BCL-XL expression. In a phase II trial in 15 patients with relapsed or refractory CLL, treatment with dasatinib (140 mg/d) achieved an overall response of 20% with progression-free survival of 7.5 months. The major adverse reaction of the treatment was myelosuppression.
SYK Inhibitors
SYK is a key protein kinase of proximal BCR signal transduction that is also involved in B-cell migration and adhesion independently of BCR activity. After BCR engagement, SYK is phosphorylated by LYN and amplifies the BCR signal through activation of downstream signaling pathways. SYK phosphorylation on the activating Y352 residue has been demonstrated in peripheral blood CLL cells. In addition, CLL cells in the lymph node show increased phosphorylation of SYK compared with peripheral blood CLL cells, indicating activation of the kinase in the tissue microenvironment.
To date, several SYK inhibitors have been studied in CLL in vitro and in vivo, including fostamatinib (R788, the oral prodrug of R406, the active metabolite), PRT318, and P505-15. In preclinical studies, treatment with SYK inhibitors resulted in inhibition of BCR activation, moderate apoptosis of CLL cells, reduced basal kinase activity of SYK, AKT, and ERK, and decreased MCL1 levels. Furthermore, SYK inhibitors have been shown to antagonize exogenous prosurvival signals provided by stromal cell or NLC coculture, secretion of BCR-regulated chemokines CCL3 and CCL4, and migration toward CXCL12 and CXCL13. In an Eμ-TCL1 transgenic mouse model, treatment with fostamatinib inhibited BCR signaling, reduced the proliferation and survival of the leukemic clone, and extended the life of the treated mice.
Fostamatinib was the first SYK inhibitor introduced into clinical study. In a phase I/II trial in patients with relapsed/refractory B-cell malignancy, fostamatinib was shown to be well tolerated with the most common adverse reactions being myelosuppression, fatigue, and diarrhea. The highest ORR was achieved in patients with CLL/small lymphocytic lymphoma (SLL) (55%, 6 of 11, with a median progression-free survival of 6.4 months), compared with only 10% to 22% in other non-Hodgkin lymphomas (NHLs). The on-target effect of fostamatinib in CLL has been demonstrated by downregulation of BCR-regulated target genes in tumor cells of patients with CLL on fostamatinib. Furthermore, fostamatinib inhibited CLL cell activation and proliferation. However, there was no correlation between the degree of inhibition of BCR signaling and clinical response suggesting that pathways bypassing BCR activation might play a role in shaping the response to such kinase inhibitors. Fostamatinib is being tested in late-stage clinical trials for rheumatoid arthritis and in patients with diffuse large B-cell lymphoma. Some novel SYK inhibitors have shown promising preclinical activity, and may have increased potency and specificity.
BTK Inhibitors
BTK is a member of the TEC family of kinases that is critical for BCR signaling. Mutations in BTK result in X-linked agammaglobulinemia, an inherited disorder manifesting with profound decrease in antibody production and a severe defect in B-cell development. BTK mRNA and protein expression levels are increased in CLL cells. Ibrutinib (PCI-32765) is an orally administered irreversible and specific inhibitor of BTK that induces modest apoptosis in CLL cells irrespective of IGHV mutational status or interphase cytogenetics, and overcomes prosurvival and proliferation signals provided by various tissue microenvironmental elements (such as CD40L, BAFF, IL-4, IL-6, TNFα, fibronectin and stromal cells coculture, and CpG oligonucleotide). Ibrutinib abrogates CLL cell signaling, migration, and adhesion in response to tissue homing chemokines (such as CXCL12, CXCL13, and CCL19) and abrogates integrin α4β1-mediated adhesion to fibronectin and VCAM-1. Both in vitro and in vivo, ibrutinib has been reported to inhibit CLL cell secretion of CLL3 and CCL4. At the molecular level, this agent inhibits BTK tyrosine phosphorylation after BCR or CD40 stimulation and abrogates activation of downstream signaling pathways including ERK, PI3K, and NF-κB in CLL cells. Treatment with ibrutinib in the TCL1 mice model of CLL resulted in inhibition of disease progression.
A phase I open-label dose-escalation study evaluated the efficacy and tolerability of ibrutinib in patients with relapsed or refractory B-cell NHL and B-cell CLL. Dose escalation proceeded to 12.5 mg/kg without dose-limiting side effects and with pharmacodynamic evidence for complete inhibition of BTK. Sixty percent of all patients achieved an overall response (OR), with 16% achieving a complete response (CR); 16 patients with CLL/SLL were evaluated, with an objective response reported in 11 of these patients (69%). The observed responses were of marked duration, as the median progression-free survival for all patients reported at the time of data cutoff was 13.6 months. A subsequent phase Ib/II study of ibrutinib in patients with CLL who were either greater than 65 years of age and previously untreated or diagnosed with relapsed or refractory disease, further demonstrated that ibrutinib was well tolerated; the most common side effects included diarrhea, fatigue, and nausea. The ORR was 71% for treatment-naive patients, 67% for relapsed or refractory patients, and 50% for high-risk patients. The estimated progression-free survival at 26 months was 75% for the relapsed/refractory cohort and 96% for treatment-naive patients demonstrating a remarkable duration of response.
Targeting the PI3K/AKT/mTOR Signaling Pathway
The PI3K/AKT/mTOR signaling pathway is a critical intracellular signaling cascade controlling cell survival and proliferation in both malignant and nonmalignant cells. PI3K is a pivotal hub connecting multiple extracellular signals to cellular responses. PI3K acts to convert phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) to (PI(3,4,5)P3), which in turn forms a functional signaling complex with BTK and AKT. Several isoforms of PI3K have been characterized; the δ isoform of PI3K is selectively expressed in hematopoietic cells and functions to relay BCR, BAFF, CD30, and TLR signaling. CLL cells express PI3Kδ and display increased PI3K activity. Given the selective expression of PI3Kδ, in contrast to the more pervasive α and β isoforms, specific inhibition of the δ isoform is not expected to be toxic to normal tissues. Many PI3K inhibitors are currently being investigated in both preclinical and clinical settings. ON 01910.Na (rigosertib), a multikinase PI3K inhibitor in phase III trials for myelodysplastic syndrome, has demonstrated promising preclinical in vitro activity, inducing apoptosis in CLL cells that are cultured in contact with stromal cells via a dual mechanism of action involving both PI3K/AKT inhibition and induction of oxidative stress.
GS-1101 (CAL-101)
GS-1101 is an orally available, highly selective PI3Kδ inhibitor that induces apoptosis in CLL cells. The cytotoxic effect of GS-1101 is maintained despite the presence of various microenviromental components that normally support malignant CLL cells (including stromal cells, NLCs, fibronectin, CD145 or TNFα BAFF and BCR stimulation). GS-1101 also inhibits the secretion of both antiapoptotic and proinflammatory cytokines. GS-1101 sensitizes CLL cells to drug-induced apoptosis in the presence of stromal coculture and inhibits CLL cell chemotaxis toward CXCL12 and CXCL13. The drug abrogates constitutive PI3K signaling in CLL cells as well as AKT and/or ERK activation by anti-IgM, soluble CD40 ligand, and chemokines. In patients with CLL treated with GS-1101, the serum levels of CCL3, CCL4, and CXCL13 were markedly reduced. A phase I study in 54 patients with previously treated CLL demonstrated the acceptable safety and promising clinical activity of GS-1101, with 26% of patients achieving an OR. A greater than 50% reduction in lymphadenopathy was observed in 80% of patients. Adverse events grade 3 or higher were minimal and included pneumonia and neutropenia; they were observed in less than a quarter of patients. Phase II/III clinical trials of GS-1101 are currently underway.
AKT Inhibitors
The serine/threonine kinase AKT is a key nodal regulator of cellular survival known to phosphorylate several cellular substrates including caspase 8, caspase 9, BAD, mTor, and the Forkhead family of transcription factors. Furthermore, AKT activation is associated with resistance to chemotherapy. Early-phase clinical trials are underway investigating the use of AKT inhibitors in several malignancies including chronic myelocytic leukemia, although experience with these agents in CLL is limited. MK-2206, an orally active allosteric AKT inhibitor, has been shown to enhance the antitumor efficacy of other chemotherapeutic agents in vitro in several malignancies, although clinical use in CLL has not been reported. Perifosine, another oral inhibitor of AKT, is undergoing phase II evaluation in patients with refractory and relapsed leukemia. Pending the outcome of these and other clinical trials, further investigation is warranted to determine the usefulness of AKT inhibitors in CLL.
mTOR Inhibitors
mTOR, a serine/threonine kinase of the PI3K/AKT/mTOR signaling network, is involved in cell growth, metabolism, and proliferation and is commonly activated in B-cell neoplasms. Rapamycin (sirolimus, rapamune, Wyeth) is an immunosuppressive drug used to prevent rejection in organ transplantation. The drug profoundly blocks BCR-mediated proliferation. Preclinical studies in CLL have shown that rapamycin or its analogue RAD001 blocks cell cycle progression by interfering with expression of critical cell cycle molecules. Everolimus (RAD001, afinitor, Novartis), an orally available derivative of sirolimus, was evaluated in a phase II pilot trial in previously treated patients with CLL. The study was stopped early because of increased toxicity, although the drug showed modest clinical activity. In another phase II study, everolimus (10 mg/d) administered to patients with recurrent/refractory CLL achieved PR in 18% of patients. In a subset of these patients, treatment with everolimus was accompanied by an increase in lymphocytosis in parallel with a reduction in lymphadenopathy.
Targeting the RAF/MEK/ERK Signaling Pathway
Sorafenib (BAY43-9006; nexavar) is an oral, small-molecule, multikinase inhibitor approved for the treatment of advanced renal cell carcinoma and unresectable hepatocellular carcinoma. Sorafenib is a potent RAF serine/threonine kinase inhibitor targeting the RAF/MEK/ERK pathway and inhibits other receptor tyrosine kinases involved in tumor progression and angiogenesis. Sorafenib induces CLL cell death that is mediated via caspase activation and a decrease in MCL1. It overcomes apoptosis protection induced by NLC or stromal cell coculture and stromal-mediated chemoresistance. The drug has been shown to block RAF/MEK/ERK signaling and the chemotaxis response induced by CXCL12 in CLL cells. It has also been shown to inhibit RAF and ERK activation by NLCs or stromal cells and to interfere with VEFGR/STAT3 signaling induced by stromal cell coculture. Sorafenib also abrogates BCR-mediated signaling and survival in CLL cells. CLL cells derived from the lymph nodes are more sensitive to sorafenib than the cells found in peripheral blood. The clinical efficacy and tolerability of sorafenib in relapsed CLL is currently being evaluated in a phase II clinical trial.
Targeting Antiapoptotic Proteins
The resistance of CLL cells to apoptosis is related to high expression of the BCL-2 family antiapoptotic proteins. The overexpression of these antiapoptotic proteins in CLL is endogenous as well as extrinsic and is in part regulated by signals derived from the tissue microenvironment. Therefore, in recent years, several therapeutic strategies have been developed to target antiapoptotic proteins in CLL, including antisense BCL-2 oligonucleotide, BH3 mimetics, and others.
Oblimersen sodium is a synthetic BCL-2 antisense oligonucleotide that induces a decrease in BCL-2 mRNA and protein levels and apoptosis in CLL cells. Oblimersen sodium, when combined with different cytotoxic drugs, increases CLL cell apoptosis. In a phase I/II trial, oblimersen sodium as a single agent, showed minimal activity in patients with relapsed/refractory CLL. Dosing was limited by development of a cytokine release syndrome. At a dosage of 3 mg/kg/d, 2 (8%) of 26 patients achieved PR. A phase III study in patients with relapsed/refractory CLL showed that addition of oblimersen to fludarabine plus cyclophosphamide (FC) increased the CR/nodular PR (nPR) rate compared with FC alone. Accordingly, CR/nPR was achieved in 20 (17%) of 120 patients in the oblimersen group and 8 (7%) of 121 patients in the FC-only group. The combination of oblimersen-FC further resulted in increased survival in subsets of patients who achieved a least a PR and in those who had fludarabine-sensitive disease.
Obatoclax mesylate (GX15-070) is a small-molecule pan-BCL-2 antagonist. This compound belongs to a class of BH3 mimetic agents that inhibit the activity of the antiapoptotic BCL-2 members that antagonize the proapoptotic proteins BAX and BAK. The BH3-only proteins BAX and BAK are directly sequestered and repressed by the antiapoptotic BCL-2 proteins. BH3 mimetic drugs BAX and BAK induce their release allowing them to oligomerize and trigger apoptosis via the formation of pores in the outer mitochondrial membrane. In a phase I trial, administration of obatoclax to patients heavily pretreated with advanced CLL resulted in a PR in 1 out of 26 patients. The major toxicities were neurologic including somnolence, ataxia, and euphoria.
BH3 Mimetics
The BH3 mimetics, ABT-737 and its orally active analogue navitoclax, inhibit BCL-2, BCL-XL, and BCL-W. In preclinical studies, ABT-737 has been shown to induce rapid and potent proapoptotic activity in CLL cells independently of the common clinical and prognostic parameters in CLL. Addition of cytotoxic agents sensitizes CLL cells in vitro to ABT-737. In a phase I study of 29 patients with relapsed/refractory CLL, a navitoclax dosage of at least 100 mg/d achieved durable PRs in 35% of patients. Navitoclax was also active in high-risk patients with fludarabine-refractory disease, bulky lymphadenopathy, and deletion of 17p. The major dose-limiting toxicity was thrombocytopenia related to inhibition of BCL-XL.
AT-101 (gossypol isomer) is a small-molecule pan-BCL-2 antagonist. In preclinical studies, AT101 was shown to both induce CLL cell apoptosis and to overcome resistance mediated by stromal cell coculture, while sparing normal stromal cells. A phase I trial of AT-101 in treatment-naive patients with CLL with high-risk disease demonstrated that the drug was well tolerated. Furthermore, 5 of 6 patients in this trial exhibited a decrease in lymphocyte count, whereas all patients demonstrated a reduction in lymphadenopathy.
XIAP
XIAP, which inhibits the proteolytic activity of caspase-3 via direct binding, is highly expressed in CLL cells, and plays an important role in TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. Because CLL cells have previously exhibited resistance to TRAIL-based treatments, novel inhibitors of XIAP have been developed in the hope of overcoming TRAIL resistance in CLL. One such novel small-molecule inhibitor, compound A (CA), has been shown to render tumor cells from patients with 17p deletion, IGVH unmutated type CLL, susceptible to TRAIL in vitro.
Immunomodulatory Drugs
Lenalidomide, a derivative of thalidomide, is an immunomodulatory agent that has significant activity in 5q− myelodysplastic syndrome, multiple myeloma, and other B-cell malignancies. In relapsed or refractory CLL, intermittent lenalidomide at 25 mg/d (3 weeks on/1 week off drug) was associated with acceptable toxicity, and achieved an ORR of 47%, with 9% of patients demonstrating CR. Another trial of relapsed/refractory patients with CLL treated with daily lenalidomide at a dose of 10 mg produced an ORR of 32%, with 7% CR. In a treatment-naive setting, elderly patients with CLL treated with lenalidomide achieved an ORR of 65%, including 10% CR and an additional 5% CR with residual cytopenias. The activity of lenalidomide is irrespective of unfavorable genomic abnormalities such as unmutated IGHV or fludarabine-refractory disease status. However, at least 6 to 9 months of treatment may be needed to achieve the best possible response. The most common toxicity of lenalidomide is myelosuppression. Lenalidomide is frequently associated with a cytokine release syndrome and tumor flare reaction (observed in 30% to 58% of patients) or tumor lysis syndrome. Tumor flare is a unique immune-mediated response to lenalidomide therapy characterized by painful lymph node enlargement that may be accompanied by fever and/or bone pain. It is generally managed with corticosteroids, although in more severe cases narcotics and hospitalization may be necessary. Tumor flare may be predictive of clinical response to treatment with lenalidomide. The optimal dosing schedule of lenalidomide is not well defined; and a dose of 25 mg once daily in cases of relapsed CLL may be associated with unacceptable toxicity in some patients. Furthermore, lenalidomide may be associated with an increased frequency of venous thromboembolism that may be related to endothelial dysfunction in the context of inflammatory cytokine secretion. Continuous low-dose lenalidomide (10 mg) is generally well tolerated and effective. The treatment is accompanied by increased serum Ig levels and typically reduces CCL3 and CCL4 plasma levels, which might indicate an inhibitory effect on intracellular signaling in CLL cells.
The mechanism of action of lenalidomide is not completely understood. In contrast to thalidomide, lenalidomide has weak antiangiogenic effects. Lenalidomide induces transient immune activation including expression of costimulatory molecules such as CD40, CD80, and CD86 on CLL cells, which may be mediated through PI3Kδ. It also increases the levels of IL-6, IL-10, IL-2R, IFNγ, and TNFα in serum. Lenalidomide in part restores T-cell function by enhancing the formation of immunologic synapses and promoting intracellular signaling. In lymph node biopsies from patients with CLL treated with lenalidomide, a shift toward a Th-1 type immune reaction with production of IFNγ was documented, suggesting that lenalidomide might be able to restore antitumor immunity and immune surveillance in vivo. Furthermore, lenalidomide enhances NK cell activity and improves antibody-mediated cellular cytotoxicity directed by rituximab.
Related posts:
 The Significance of Stereotyped B-Cell Receptors in Chronic Lymphocytic Leukemia
The Significance of Stereotyped B-Cell Receptors in Chronic Lymphocytic Leukemia
 The Role of Minimal Residual Disease Measurements in the Therapy for CLL
The Role of Minimal Residual Disease Measurements in the Therapy for CLL
 The Evolving Role of Stem Cell Transplantation in Chronic Lymphocytic Leukemia
Biology of Chronic Lymphocytic Leukemia in Different Microenvironments
The Significance of Stereotyped B-Cell Receptors in Chronic Lymphocytic Leukemia
Chimeric Antigen Receptor Therapy for Chronic Lymphocytic Leukemia
The Evolving Role of Stem Cell Transplantation in Chronic Lymphocytic Leukemia
Biology of Chronic Lymphocytic Leukemia in Different Microenvironments
The Significance of Stereotyped B-Cell Receptors in Chronic Lymphocytic Leukemia
Chimeric Antigen Receptor Therapy for Chronic Lymphocytic Leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


