Biology of Aging and Longevity: Introduction
Aging is the process that converts young adults, most of them healthy and in no need of assistance from physicians, into older adults whose deteriorating physiological fitness leads to progressively increasing risks of illness and death. The effects of aging are so familiar to health professionals and aging adults that it is viewed by both parties as something immutable, taken for granted, an arena in which diseases and their treatments take place, but not itself subject to intervention or modulation. The major discovery in biogerontology, gradually emerging from decades of work in animal model systems, is that this old-fashioned viewpoint is wrong, and that the aging process can be delayed or decelerated in mammals, built very much like human beings, by simple manipulations of nutritional signals and genetic circuits similar to those already well-documented in people. It is now absolutely routine to extend lifespan, in rats and mice, by about 40%, i.e., about 10 times the increase in active life expectancy that would ensue from a complete elimination of all neoplastic illnesses, or all heart attacks, in a human population. It thus seems plausible that a more detailed understanding of the factors that determine aging and the processes by which aging increases the risk of such a wide range of lethal and nonlethal illnesses and disabilities could, in the foreseeable future, have a profound impact on preventive medicine.
Aging is a mystery, in the same sense that infectious disease was once a mystery, and consciousness still is: an area of investigation in which well-informed researchers cannot really be confident that they have selected a line of investigation bound to be productive. Until recently, most published papers in biogerontology journals consisted of descriptions of the ways in which young mice, rats, or people differed from older ones, originally in terms of anatomy and physiology, as indicated by levels of enzymes or hormones, and more recently by exhaustive catalogs of protein and mRNA levels. This descriptive era is gradually being superseded by one focused on specific molecular hypotheses about the key factors that regulate aging. The foundation of this modern approach to the biology of aging was the development of genetic models and nutritional manipulations which could delay aging, and the exploitation of this leverage to test molecular ideas about the basis for the retardation of the aging process.
What Is Aging?
This question—what is aging?—is posed not as an invitation to semantic quibbling, but to initiate reexamination of facts so familiar that they are seldom examined. A case history of an individual who has mild arthritis, some loss of hearing acuity, some evidence of incipient cataract, loss of muscle mass and strength, a progressive decline in capacity for aerobic exercise, troubles with learning and remembering, and an increased vulnerability to infectious illness would lead any physician to assume that the individual described is a man or woman of 60 or more years of age. But the list of signs and symptoms refers with equal accuracy to a 20-year-old horse, or a 10-year-old dog, or a 2-year-old mouse. The specific list of deficits and impairments shifts a bit from person to person, and from species to species, but it is extremely rare to find an 80-year-old person, or 3-year-old mouse, or 14-year-old dog that has avoided all of these age-associated problems. The aging process is synchronized, in that it is common to see all of these difficulties in older people, horses, dogs, and mice, but rare to encounter any of them in young adults just past puberty. This synchrony, though entirely familiar to schoolchildren, physicians, and scientists alike, is the central challenge in biological gerontology: how is it that such a process affects so many cells, tissues, organs and systems at a rate that varies, even among mammals, over a 100-fold range from the shortest lived shrews to the longest lived whales? Structural features of shrews, mice, dogs, and people are remarkably similar at scales from the arrangement of DNA and histones in the nucleus, to the architecture of the kidney, heart, and thymus, to the role of the central nervous system (CNS) and endocrine systems in regulating responses to heat and cold, hunger and thirst, infection, and predators. Why, then, in molecular terms, will the eye, kidney, immune system, brain, and joints of a mouse last only 2 to 3 years under optimal conditions, while the same cells and organs and systems persevere for 50 or more years in people, and longer still in some species of whales?
The definition proposed at the beginning of this chapter—aging as a process for turning young adults into distinctly less healthy old ones—is straightforward enough to appear simple-minded, but in practice draws some prominent distinctions. In this view, aging is not a disease: Diseases are certainly among the most salient consequences of aging, but aging produces many changes not classified as diseases, and many diseases also affect young people. Similarly, lifespan and mortality risks are influenced by many factors besides aging. Thus, evidence that a gene or diet or public health measure has altered life expectancy, upwards or downwards, does not imply that the effects have been achieved by an effect on aging. In the context of the whole organism definition of aging used in this chapter, it is hard to interpret the meaning of changes that occur as individual cells “age” in tissue culture. While cell culture studies can provide valuable information of great relevance to ideas about aging, the two processes are likely to be fundamentally different.
From a biological standpoint, a critical distinction is the difference between aging and development. Development creates a healthy young adult from a fertilized egg, and is strongly molded by natural selection. Genetic mutations that impair development, creating a slower falcon, a near-sighted chipmunk, or a chimpanzee uninterested in social cues, are rapidly weeded from the gene pool. But the force of natural selection diminishes dramatically at ages that are seldom reached. Mice, for example, typically live only 6 months or so in the wild, before they succumb to predation, starvation, or other natural hazard. There is strong selective pressure against mutations that cause cataracts in the first few months of mouse life, but little or no pressure against genotypes that postpone cataract formation for 2 years. Mice protected, in a laboratory setting, against predation, starvation, and other risks typically do develop cataracts in their second or third year. In wolves, however, a genotype that delayed cataracts for only 2 years would be a disaster; natural selection favors wolf genes that preserve lens transparency for a decade or more. A similar process, working on our ancestors in environments where survival to 15 was common, but survival to 55 distinctly uncommon, has filled our own genome with alleles that postpone cancer, osteoarthritis, coronary disease, Alzheimer’s, presbycusis, cataracts, sarcopenia, immune senescence, and many other familiar maladies, for about 50 to 60 years. Thus, although aging and development seem, superficially, to be similar processes in that both lead to changes in form and function, they are different in a fundamental and critical way: Development is molded directly by the forces of Darwinian selection and the changes of aging are the consequence of the failure of these selective processes to preserve function at ages seldom reached by individuals in any given species.
The definition of aging as a process that turns young adults into old ones conflicts with a view of aging as instead a collection of processes, some that lead to arterial disease, some that affect endocrine function, some that impair cognition or cause neoplastic transformation, etc. Because each of these ailments is itself the outcome of a complex interaction among many factors, including genes, diet, accidents, viruses, toxins, antibiotics, and physicians, and because each of these diseases, and many others, seems an inextricable part of aging, it has seemed implausible to regard aging as less complex than its (apparent) constituents. Considering aging as a process, rather than a collection of complex processes, has thus seemed to be an oversimplification.
However, two lines of evidence support the merits of the view of aging as a unitary process, with its own (still ill-defined) physiological and molecular basis, which underlies and tends to synchronize the multiple changes seen in older individuals. The first of these discoveries was caloric restriction: The observation that rodents allowed to eat only 60% of the amount of food they would voluntarily consume would live 40% longer than controls permitted free access to food. This observation, first made by McCay in the 1930s, has now been repeated in more than a dozen species in scores of laboratories, and ongoing studies in rhesus monkeys have now produced preliminary, but highly suggestive, evidence that similar benefits may accrue in our own order of mammals. The key point is not merely that lifespan is extended, but that nearly all of the consequences of aging are coordinately delayed. Caloric restriction delays changes in cells that proliferate continuously (such as gut epithelial cells), cells that can be triggered to proliferate when called upon (such as lymphocytes), and those that never proliferate (such as most neurons), as well as on tissues that are extracellular or acellular (such as lens tissue and extracellular collagen fibrils). It delays aspects of aging characterized by excess proliferation, such as neoplasias, and those characterized by failure to proliferate (such as immune senescence). It delays or decelerates age change at the tissue level (such as degradation of articular cartilage) and those involving complex interplay among multiple cells and tissues (such as loss of cognitive function and endocrine control circuits). Because caloric restriction alters, in parallel, so vast an array of age-associated changes, it seems inescapable that these many changes, distinct as they are, must be in some measure timed, i.e., synchronized by a mechanism altered by caloric intake.
A second, more recent, set of experiments leads to the same inference. In 1996, Bartke and his colleagues showed that Ames dwarf mice, in which a developmental defect in the pituitary impairs production of growth hormone (GH), thyrotropin, and prolactin, had an increase of more than 40% in both mean and maximal lifespan compared to littermates with the normal allele at the same locus. Since then studies of this mutant, and the closely similar Snell dwarf, have documented delay in kidney pathology, arthritis, cancer, immune senescence, collagen cross-linking, cataracts, and cognitive decline, making a strong case that these genetic changes in endocrine levels do indeed modulate the aging process as a whole, with consequent delay in a very wide range of age-synchronized pathology. Since 1996, mouse researchers have documented increased lifespan in at least nine other mutations, of which five others, like the Ames and Snell mutations, lead to lower levels of or responses to GH and/or its mediator insulin-like growth factor I (IGF-I).
These observations, on calorically restricted (CR) rodents and now also in mutant mice, justify a sea change in thinking about aging and its relationship to disease. The new framework includes two key tenets: (1) the aging process, despite the complexity of its many effects, can usefully be considered as a single, coordinating mechanism and (2) the rate at which aging progresses can be decelerated in mammals as well as in other taxa. From this perspective, a fundamental challenge to biogerontologists is to develop and test models of how age-dependent changes are themselves coregulated, during middle age, to produce old people or old mice or old worms. Studies of the aged, as opposed to studies of aging, are relevant to this challenge only insofar as they are exploited to generate or test ideas about coregulation and synchrony, rather than ideas about disease-specific pathogenesis. The key resource for meeting the challenge is not comparisons between young and old donors, but rather comparisons between young or middle-aged adults who are known to be aging at different rates. Fortunately, the same experiments that have documented the malleability of aging rate have done so by producing sets of animals that do indeed age at normal or slower-than-normal paces. With luck, future studies may produce at least tentative answers to the two key questions in biogerontology: How does aging produce the signs and symptoms of aging and what controls the rate of this process in mammals?
Four main ideas have emerged, in the period 1990–2008, from studies of genetically convenient invertebrate models, the nematode worm Caenorhabditis elegans and the fruit fly Drosophila melanogaster. (1) Single gene mutations—lots of them—can extend lifespan in worms and flies (and mice). Most mutations discovered so far are “loss of function,” in which eliminating or crippling the gene product leads to slower aging and lifespan extension. (2) The genes whose elimination slows aging are typically those used by the normal animal to notice and respond to environmental poverty; they are selected, by evolution, to permit the animal to take on alternate forms or functions to deal with the relative absence of nutritional conditions optimal for rapid growth and reproduction. Many of these genes are familiar to physicians and biological scientists: they encode insulin, insulin-like growth factors, possibly thyroid hormones, and other regulators of fuel usage and metabolic fluxes, as well as the intracellular proteins that change cell properties in response to these hormones. (3) The mutations that extend lifespan in worms and flies (and perhaps in mice) render the animals more resistant to lethal injury, such as that resulting from heavy metals, ultraviolet irradiation, heat, oxidizing agents, and chemicals that damage DNA. It thus seems plausible that augmentations of stress resistance are the mechanism by which these mutant genes slow aging. (4) This three-way association, connecting aging to stress to signals about nutrition, has very deep evolutionary roots and can be noted (albeit with species-specific nuances) in yeast, flies, worms, and mice. The suggestion here is that the pathways tying nutrition to stress to aging rate evolved extremely early in the eukaryotic lineage, in an ancestor predating the branch points between yeast, flies, worms, and vertebrates. The key implication is that investigations into the cell biology of these linked processes in conveniently short-lived organisms may provide valuable insights into aging and disease in humans.
Studies of the invertebrate model systems have also called attention to a range of intracellular pathways that influence longevity, perhaps through regulation of resistance to a variety of forms of lethal and metabolic stress. Components of these pathways are now under scrutiny to see if they also affect aging and disease in rodents. Many of the antiaging mutations in worms, for example, act by increasing the actions of a DNA-binding factor called Daf-16, whose targets include genes that modulate resistance to oxidative damage, DNA repair, and other modes of cellular defense. The mammalian equivalent to Daf-16, a member of the FoxO group of transcription factors, has already been implicated in control of cell death in human and rodent cells, and studies of the role of FoxO family members in aging are under way. A second family of proteins, the sirtuins, was initially implicated in lifespan regulation in studies of the budding yeast Saccharomyces cerevisiae, but then shown also to be able to increase lifespan in nematode worms. There is some evidence that members of the sirtuin family may contribute to the improved health and longevity of mice on a calorie-restricted diet, and interest in these proteins has been spurred by observations that median lifespan of mice on a diet high in saturated fat can be increased significantly by resveratrol, whose many biochemical effects include stimulation of sirtuin activity. Work with invertebrates has also brought new attention to the possible role of TOR, the “target of rapamycin,” in control of lifespan and responses to dietary interventions. TOR plays a major role, conserved throughout the evolutionary tree, in regulating protein translation rates in response to both external cellular stress and the available supply of amino acids. TOR inhibition by rapamycin is a mainstay of clinical immunosuppressive therapy after organ transplantation, and TOR inhibitors also show promise as antineoplastic agents. Other molecular circuits, including those triggered by the p66 (shc) protein, the tumor-suppressor p53, and the stress-responsive kinase JNK, were initially discovered through work with rodents and now are also being examined in the context of invertebrate models for aging. Each of these biochemical pathways has powerful and still only partially defined links to hormonal signals, neoplastic transformation, stem cell homeostasis, and the balance between cell growth and cell death, so their elucidation is likely to provide both rationale and direction for translational work aimed at slowing the aging process and retarding age-related disease and dysfunction.
Mice or rats fed approximately 30% or 40% less food than they would ordinarily consume typically live up to 40% longer than freely fed animals. These CR animals stay healthy and active, with good physical, sensory, and cognitive function, at ages at which most of the controls have already died. The intervention extends lifespan if initiated at very young ages (e.g., at weaning), or when started early in adulthood (e.g., at 6 months, in a species where puberty occurs at 2 months and median survival is about 28 months). Whether CR diets extend lifespan when initiated in animals already older than half the median lifespan is controversial, with some early studies suggesting little or no response, and some more recent experiments leading to a positive result. Lifespan can be extended using CR diets of widely varying composition, and there is strong evidence that total caloric intake, rather than proportions of specific fuel sources (carbohydrates, fats, proteins), is responsible for the beneficial effects. CR diets do not, except in the first few months after their imposition, alter metabolic rate or oxygen consumption per gram of lean body mass because CR rodents lose weight, or if adolescent fail to gain weight, so that lean body mass matches calorie supply once equilibrium is established. Although CR rodents are less obese than control animals, it seems unlikely that the effect of CR diets on aging is caused entirely by avoidance of obesity, in part because CR diets extend lifespan in mice genetically engineered to lack leptin (ob/ob mice). When placed on a CR diet, ob/ob mice are both longer lived and more obese than normal mice with normal caloric intake. There is some evidence that CR diets can delay at least some aspects of aging, for example, changes in immune function, in nonobese rhesus monkeys, but definitive evidence, based on lifespan data, is not yet available.
Mice and rats on CR diets show delay or deceleration in a very wide range of age-dependent processes, including neoplastic and nonneoplastic diseases, changes in structure and function of nearly all tissues and organs evaluated, endocrine and neural control circuits, and ability to adapt to metabolic, infectious, and cardiovascular challenges. Mouse stocks that have been engineered for vulnerability to specific lethal diseases, such as models of lupus or early neoplasia, also tend to live longer when placed on a CR diet. Intensive study of CR rodents in the past 20 years has suggested many ideas about the mechanism of its effects, including ideas about altered levels and responses to glucocorticoids and/or insulin, increases in stress-resistance pathways including resistance to oxidative injury, diminished inflammatory responses, changes in stem cell self-renewal, and many others, each plausible, none of them at this point more than plausible. Further work on the early and midterm effects of CR diets is among the most attractive avenues for testing basic ideas about aging in mammals.
Rats fed a diet containing much reduced levels of the essential amino acid methionine were shown, in 1993, to live 40% longer than rats, on a standard diet, and more recent work has shown a similar, though smaller, effect on mice. These animals are not calorically restricted—they eat more calories, per gram of lean body mass, than control rats, and rats pair-fed normal food at levels that match the total caloric intake of a methionine-restricted (MR) rat show a much smaller degree of lifespan extension. MR diets could, in principle, affect aging by causing changes in protein translation or rate of protein turnover, by changes in DNA methylation (which depends upon metabolites of methionine), by alterations in levels or distribution of the antioxidant glutathione (also a metabolite of methionine), by changes in hormone levels (MR mice show low levels of insulin, glucose, and insulin-like growth factor 1, IGF-I), or by induction, through hormesis, of augmented stress response pathways at the cellular level. Some of these ideas could be tested using diets that restrict levels of other amino acids, and there is older, fragmentary evidence that such diets, too, may induce lifespan extension in rodents. MR is thus the second confirmed method for extending lifespan in mammals and comparisons of similarities and differences between CR and MR rodents are likely to prove highly informative.
The initial reports in the 1980s and 1990s that mutations of single genes in worms, and then in flies, could produce dramatic increases in lifespan were the strongest support, along with the CR data, for viewing aging as a unitary process that could be decelerated. The report in 1996 by Bartke and his colleagues that the lifespan of mice could be extended more than 40% by mutation of a gene required for pituitary development opened the door to new genetic models for study of aging in mammals. This Ames dwarf gene (Prop1) leads to an endocrine syndrome featuring low levels of growth hormone (GH), IGF-I, thyrotropins and the thyroxines, and prolactin. The observation that longevity in mice could be improved by reduction of hormones in the insulin/IGF family, i.e., the same family of signals implicated in lifespan extension in worms and (later) flies, provided a key foundation for exploiting comparative cell biology in biogerontology research. Subsequent work then showed that similar degrees of lifespan extension could be seen in Snell dwarf mice, whose endocrine defects are very similar to those of Ames dwarfs, and also in mice that lack the GH receptor (GHR-KO mice) (Figure 1-1). This last observation, together with documented lifespan extension in mice lacking GH-releasing hormone receptor (GHRHR) and in mice with diminished expression of the IGF-I receptor (IGF1R heterozygotes), suggested strongly that a common factor, i.e., low production of or response to IGF-I, is a major cause of lifespan extension in all five models, although it is certainly possible that other factors, such as altered insulin sensitivity, thyroid tonicity, adipokine levels, etc., may contribute to antiaging effects in some of these mouse models. Male and female mice are affected by each of these mutations except that the effect of the IGF1R mutation seems stronger in females than in males. Mice in which tissue levels of IGF-I are reduced by genetic manipulation of a protease that controls local concentrations of IGF-I binding proteins are also long-lived, again consistent with models in which abnormally low IGF-I levels cause lifespan extension in mice. Studies of Snell and Ames dwarf mice have shown that the exceptional longevity of these mice is accompanied by a delay or deceleration of age-dependent changes in T lymphocytes, skin collagen, renal pathology, lens opacity, cognitive function, and neoplastic progression; taken with the lifespan data, these observations suggest strongly that these mutations, like the CR diet, act to slow the aging process itself.
Figure 1-1.
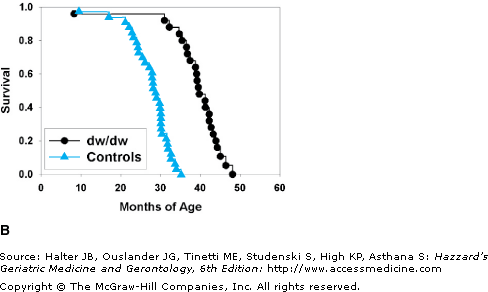
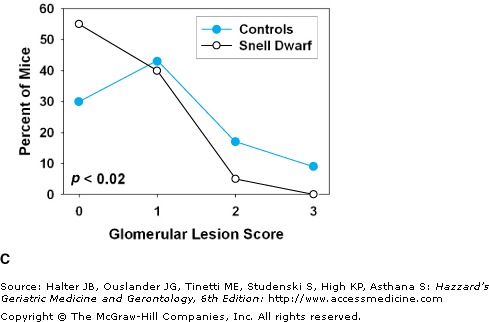
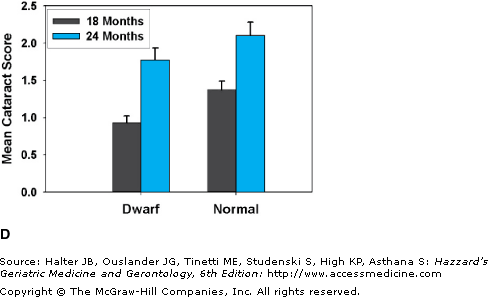
(A) A young adult Snell dwarf mouse, with a littermate control (on the vehicle). (B) Survival curves for Snell dwarf (dw/dw) mice and littermate controls. (C) Glomerular basement pathology scores, at terminal necropsy, for Snell dwarf (N = 40) and control (N = 46) mice. Higher scores indicate a greater degree of kidney pathology. Despite living 40% longer, a higher percent of Snell dwarfs had 0 scores at necropsy and a smaller percent had scores of 2 or 3. (D) Cataract scores determined by slit lamp examination in Snell dwarf and littermate control mice at 18 and 24 months of age. p < 0.001 for the difference between dwarfs and controls at each age. (B) Data from Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A. 2001;98(12):6736–6741. (C and D) Data from Vergara M, Smith-Wheelock M, Harper JM, Sigler R, Miller RA. Hormone-treated Snell dwarf mice regain fertility but remain long-lived and disease resistant. J Gerontol Biol Sci. 2004;59:1244–1250.
In addition to the six mutations that lead to lower levels of IGF-I signals, there are now five other mutations that have been shown, in each case in a single unreplicated report, to extend lifespan in mice. Table 1-1 presents a summary of these 11 published mutations that extend maximal lifespan in mice. Transgenic overexpression of urokinase-type plasminogen activator in the brain has been reported to produce a significant extension of lifespan, perhaps by suppression of appetite with consequent mimicry of a CR diet. Mice in which the insulin receptor has been inactivated specifically in adipose tissue show an 18% lifespan increase, consistent with the idea that altered insulin sensitivity or adipokine levels might play a role in aging rate in this species. Transgenic mice overexpressing the klotho protein, a cofactor for fibroblast growth factor (FGF) signals whose absence leads to early death by elevation of vitamin D levels, have been reported to live 19% to 31% longer than controls, perhaps reflecting the ability of klotho to block insulin or IGF-I signals. A mutation that inactivates the 66 kDa splice variant of the Shc protein, involved in the pathway to programmed cell death after exposure to hydrogen peroxide or ultraviolet light, also extends longevity (by about 28%), as does transgenic overexpression, in mitochondria, of catalase, an enzyme involved in detoxification of hydrogen peroxide (a 20% lifespan increase). These last two reports, if confirmed in other laboratories, may give new insights into the connections linking aging and late-life diseases to agents that damage DNA or induce oxidative injury at intracellular sites.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


