Fig. 3.1
Hypothetical explanations why there is no primary lesion detectable in CUP. See text for details (Figure adapted from Löffler et al. [4])
The primary tumor is present, but not recognized as being the primary because of atypical histologic features, e.g., a lung lesion with features of gastrointestinal adenocarcinoma. Quite frequently, the distinction between CUP and a defined primary is arbitrary, as, for example, in case of CK7-positive adenocarcinoma with one prominent liver lesion, which could be intrahepatic cholangiocarcinoma as well as a metastasis from a primary somewhere else within the upper gastrointestinal tract; these possibilities cannot be reliably distinguished from each other by immunohistochemistry [5]. Sometimes, rare cancer types are missed to consider. An example is adenocarcinomas presenting as solitary skin lesion, which are quite frequently classified as CUP but could as well be primary adnexal carcinomas of the skin [6]. In such cases, it is advisable to perform a thorough search for primary lesions located somewhere else and, whenever there is no distant primary detected, to drop the CUP diagnosis in favor of the assumption that the solitary (or leading) lesion is in fact the primary.
A related situation is the malignant transformation of ectopic tissue which is usually not present at the site of origin. This seems to be a major underlying cause of extragonadal germ cell tumors, which are therefore classified as germ cell tumors and not as CUP if the germ cell phenotype is obvious [7]. This assumption is largely based on the fact that there are benign counterparts – teratomas – which are known to occur also at extragonadal sites [8]. If the germ cell phenotype of a malignant tumor is not obvious, a germ cell tumor might be misclassified as CUP. Hence, CUPs with midline distribution and germ cell features are to be treated as germ cell tumors, as discussed in greater detail in later chapters.
The primary tumor has been accidentally removed before it became evident, but after having seeded metastases. For example, we have noticed that patients with CUP compatible with genitourinary tract primaries quite often present with a history of genitourinary tract surgery or related interventions such as hysterectomy or cervical conization, which sometimes had revealed suspicious findings such as in situ neoplasms. One might object that such interventions are performed very frequently in general, so in order to prove an association between CUP and preceding resections of occult primary lesions, it would be necessary to investigate this correlation in a systematic study, which has however, to our knowledge, not been performed yet.
The primary tumor has disappeared. This may be the extreme variant of what will be discussed later on, namely, a growth inhibition of the primary lesion, which in this case not only has stopped growing but even shrinked until extinction. For example, it has been reported that testicular cancers can present as metastases with a “burned-out” primary tumor, consisting of a fibrous scar, often associated with intratubular germ cell neoplasia [9]. Some authors believe that “burned-out” primary tumors are the mechanism underlying extragonadal germ cell tumors [9], thereby providing an alternative hypothesis to the above-discussed model of malignant transformation of ectopic tissue.
The primary lesion has spread to one or several metastatic sites before becoming malignant. This conclusion may be drawn from a study of squamous cell CUPs involving cervical lymph nodes, which used microsatellite analysis to search for the primary site [10]. In 10 of 18 patients studied, a histologically benign mucosal biopsy specimen showing a pattern of genetic alterations identical to that present in the lymph node metastases was found [10]. This is a somewhat counterintuitive finding since it indicates that a clonal population can metastasize before it acquires other features of malignancy such as invasive growth, thereby inverting the traditional model of the adenoma-carcinoma sequence placing metastatic spread at the end of tumor evolution.
Having listed these possible exceptions, we come back to the general view that most CUPs likely reflect a selective growth advantage of metastases or a growth inhibition of the primary. These two possibilities may not be exactly identical, since the former assumes that the metastases simply grow faster than the primary, which might grow slowly or not at all but is not actively inhibited, while the latter possibility might include active inhibition of the growth of the primary, which might eventually even be eradicated, as discussed above. We will discuss possible underlying mechanisms later in this chapter, but first have a look at some basic facts that may help us understand the biology of CUP.
3.3 A Short Overview of Methodology Used in Studies on CUP Biology
Here, we cannot provide a comprehensive coverage of all methods used in the context of tumor biology research. Instead, we would like to point out how research on the biology of CUP has evolved historically.
The oldest approach toward CUP biology were autopsy studies, some of which were published in the 1970s, with the oldest autopsy dating back to 1944 [3]. The same approach – to search for the primary lesion by histomorphologic examination of the putative site of origin – was later also applied to living patients, e.g., by examining resected testicles in cases with extragonadal germ cell tumors [9]. Other methods with longer tradition include cytogenetic studies, Southern blot, and fluorescence in situ hybridization [11]. Finally, the first relevant experiments using model organisms such as mice also date back to the 1970s [12].
The description of oncogenes and tumor suppressor genes in the 1970s and the clinical implementation of immunohistochemistry in the 1980s prompted the use of this method to study the tumor cell-specific expression of relevant genes since the 1990s [13]. Also starting in the 1990s, mutations of relevant genes were investigated by sequencing [14]. In the era of Sanger sequencing, this implied not only selection of genes but typically also selection of relevant exons to be examined, hence requiring educated guesses on where relevant mutations might be found.
The general limitation that not more than a few genes at a time could be examined was overcome by array-based technologies such as gene expression and array comparative genomic hybridization, enabling researchers to study large numbers or even all relevant genes concomitantly. The first studies utilizing these methods to study CUP were published shortly after 2000 [15]. During the following decade, gene expression profiling was established as a method to assign likely sites of origin to CUP specimens by comparing their gene expression profiles with the profiles of tumors of defined origin. Hence, gene expression profiling is nowadays regarded as a promising clinical tool which may be useful to guide therapeutic decisions. This will be discussed in greater detail in Chap. 11. On the other hand, these studies so far have not contributed to our understanding of CUP biology, partly because they were not designed for that purpose and partly because mRNA expression data alone are not sufficient to explain pathogenetic mechanisms.
The latest improvement of the repertoire of methods to study CUP biology is massively parallel sequencing, also referred to as next-generation sequencing, which can be applied in two variants: Either the whole genome or exome of a tumor specimen is sequenced, which requires parallel analysis of healthy tissue to distinguish tumor-specific somatic mutations from germline variants, or sequencing is restricted to a panel of relevant genes, which reduces the complexity of analysis. A pioneering study applying next-generation sequencing to CUP cases was published in 2013 [16]. Since several meeting abstracts on this topic have been published, it is safe to assume that more publications can be expected over the next few years, which will clearly scale up our knowledge about the genetic basis of CUP. It remains to be seen what this will tell us about the biology of CUP.
3.4 Sites of Origin of CUP
Once the diagnosis of CUP has been established, it is very unlikely that a primary tumor becomes evident during lifetime. According to own experience, this is the case in less than 10 % of patients – provided that the diagnosis of CUP has been based on an appropriate staging. Interestingly, we failed to find any systematic evidence (i.e., a prospective study) regarding this relationship. It seems that if the growth of a primary lesion is inhibited or has stopped, it usually does not resume growth during the course of the disease. Hence, one is left with autopsy data. A review of existing studies revealed 12 studies comprising 884 autopsies performed within the period from 1944 to 2000 [3]. The most frequently identified site of origin was the lung (20 %), followed by the pancreas (17 %). Other gastrointestinal sites were identified in 15 % of cases. Interestingly, in total a primary was identified in 73 % of cases, while it remained undetected even after autopsy in 27 % of cases (Fig. 3.2), raising some doubts about whether there is always a primary lesion, as discussed above in this chapter already. This study also included a comparison between autopsies performed before and after 1980, which did not reveal major differences, neither regarding the frequencies of specified primary sites nor with regard to the overall rate of identified versus unidentified primaries [3].
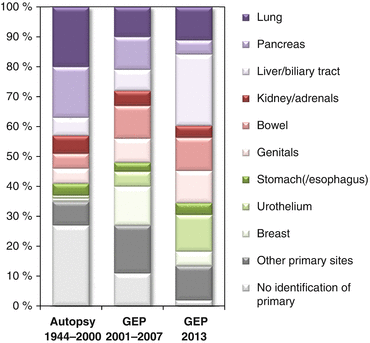
Fig. 3.2
Relative distribution of sites of origin of CUP assigned by different methods. Left: Pooled data from 844 autopsies performed between 1944 and 2000 [3]. Middle: Pooled data from gene expression profiling (GEP) of more than 500 cases from four studies published in the years 2001–2007 [3]. Right: GEP data of 252 patients from a study published in 2013 [17] (Figure adapted from Löffler et al. [4])
The same publication also investigated the frequency distribution of primary sites assigned by gene expression profiling and related methods, taking into account four studies with more than 500 cases published between 2001 and 2007 [3]. Surprisingly, this revealed major differences to the autopsy data (Fig. 3.2): With gene expression profiling, the breast was the most frequent primary organ (13 %), while a breast primary was a rare finding in autopsies (less than 1 %). Other frequent molecularly assigned primary sites were the pancreas (11 %), bowel (11 %), and lung (10 %), which was more similar to what was expected from the autopsy data. To further complicate matters, a large study was published in 2013 which included 252 patients where gene expression profiling was successfully performed to direct site-specific therapy [17]. Within that cohort, the most frequently assigned primary site was the biliary tract (21 %), which contradicted both the autopsy findings (liver or bile duct identified as primary sites in 6 % of cases) and the older gene expression data published between 2001 and 2007 (liver or bile duct assigned as primary site in 7 % of cases; Fig. 3.2). The second most frequent primary site according to the 2013 data was the urothelium (12 %), which was in agreement with the 2001–2007 gene expression data (5 % bladder/ureter) but not with the autopsy findings (1 % bladder/ureter). Other frequent primary sites in the 2013 series were the colorectum (11 %) and non-small cell lung cancer (11 %), while the pancreas accounted for only 5 % of molecularly assigned primary sites in this latest series [17].
How can these discrepancies be explained? One point that needs to be considered is that the distribution of primary sites might have changed in recent years due to changed epidemiology and due to the fact that improved diagnostic possibilities might have reduced the overall fraction of cancers classified as CUP. One might object that changes in epidemiology were not apparent in the comparison between older and newer autopsy data. Another confounder may be differences in definitions such as the fact that in the autopsy series, primary sites were defined as physical organs (e.g., liver comprising different histologies such as hepatocellular and cholangiocellular carcinoma), while gene expression profiling assigns histological types (e.g., urothelium comprising different physical sites such as bladder, ureter, renal pelvis), but a closer look at the data seems to indicate that this cannot explain all discrepancies, e.g., regarding the breast as primary site. A third confounder may be patient selection, since gene expression profiles have been mostly studied in the setting of tertiary/quaternary referral centers, as detailed in Sect. 2.2 of this book. Also taking into account this, we still doubt that all discrepancies regarding frequencies of primary sites can be readily explained. We therefore should keep in mind that gene expression-based site of origin assignment has never been verified by autopsy data. Instead, its verification was based on its ability to correctly classify tumors with known primary origin on one hand and the concordance with immunohistochemical and clinical suspicions on the other hand. With regard to the biology of CUP, an interesting alternative to explain these discrepancies would be molecular and phenotypic mimicry, i.e., tumors originating from a given organ change their phenotype (and the corresponding gene expression profile) in a way resembling the profile of another organ. This might also explain why sometimes histologic types obviously contradicting the clinical distribution pattern of metastatic spread are found. Of course, this mimicry hypothesis is speculative, but the methods described above used to verify gene expression-based assignment would be blind toward such mimicry. After all, it would be highly desirable to systematically verify molecularly assigned sites of origin in autopsied patients.
3.5 Chromosomal Aberrations, Aneuploidy, and Tumor Evolution in CUP
A classical hallmark of cancer is chromosomal instability facilitating tumor evolution [18]. Chromosomal instability leads to aneuploidy, and hence aneuploidy is often taken as an indicator of underlying chromosomal instability [19], although it has to be mentioned that aneuploidy is defined as a state (a numerical deviation from the diploid karyotype), while chromosomal instability is defined as a rate (the frequency of changes of chromosome content per cell division, often measured as variation of the number of chromosomes in a given population). Indeed, aneuploidy is found in the majority of solid cancers [19, 20]. As early as 1985, it was published that among 152 patients with metastatic adenocarcinoma of unknown primary site, 70 % showed aneuploidy detected by flow cytometric measurement of DNA content [21]. Although there were no major differences in overall survival, the authors pointed out that 18 % of patients with diploid tumors but only 9 % of patients with aneuploid tumors survived for more than 2 years [21].
A more recent study [22] combined microsatellite analysis with immunohistochemistry to compare different metastatic lesions from 14 CUP patients. In four of these cases, this was complemented by comparative genomic hybridization (CGH). The authors could not only verify a clonal relationship between multiple lesions present in each patient, but also presented evidence for clonal evolution at least in a subset of cases [22].
Several groups have characterized CUP on the chromosomal level by classical cytogenetics, CGH, or fluorescence in situ hybridization (FISH) [23, 24]. Apparently, CUP is a cytogenetically heterogeneous disease without high percentages of recurrent chromosomal aberrations. As possible application, it has been demonstrated that the cytogenetic profile is suitable to pinpoint to an organ of origin in a fraction of cases: One study published in 2003 demonstrated that 5 out of 20 patients originally classified as CUP could be reclassified by means of G-banding, CGH, and FISH, 5 of whom had lymphoma [24]. However, it should be noted that with current immunohistochemical methodology, it might be less likely that lymphomas are misclassified as CUP. Another interesting finding of this study was that a lower number of chromosomal changes (up to five versus more than five) were associated with a significantly better survival [24].
A more specific role has been established for gains of the short arm of chromosome 12, most typically as isochromosome i(12p), which is a typical aberration of testicular germ cell cancer [25]. In 1995, Motzer et al. [11] published a series of 40 CUP cases, of whom 12 (30 %) displayed an i(12p), an increased 12p copy number, or a deletion of the long arm of chromosome 12. This patient group likely suffering from germ cell tumors showed a response rate of 75 % to cisplatin-based therapy, whereas in patients without these cytogenetic findings, the response rate to cisplatin was only 18 % [11]. Similar results were published by others [26]. Hence, tumors with i(12p) or equivalent findings should be treated as germ cell tumors, and cytogenetic studies are suited for identification of such cases. We will come back to CUP with germ cell features in later chapters since this is one of the established prognostically favorable subgroups.
3.6 Genetic Aberrations in CUP Tumors
Several publications addressed genetic aberrations in CUP in the era before the advance of high-throughput technologies. This implied the directed investigation of known oncogenes and tumor suppressors, thereby aiming to reproduce the findings in larger entities such as lung or colorectal cancer. Compared to these entities, study populations of CUP patients were typically small and heterogeneous, with different publications being less well comparable to each other due to differing standards for definition of CUP. In addition, the applied methods vary from study to study, as do the proteins and genes examined, making it difficult to systematize results. In the following, we have tried to arrange the discussed publications mainly according to protein(s) and pathway(s) involved.
One of the first studies of oncogene expression in CUP samples was published in 1995 [13]. Among 26 patients, 96 % were judged positive for MYC, 92 % for RAS, and 65 % for HER-2/neu expression. This was based on immunohistochemistry staining, which impedes reliable conclusions about a true overexpression resulting from genetic changes. Hainsworth et al. [27] used the standardized DAKO score to investigate HER-2/neu expression in 94 CUP samples, among which 10 cases (11 %) showed HER-2/neu overexpression (at least 2+ according to DAKO score). Similar results were obtained in another independent investigation [28], reporting HER-2/neu overexpression in 16 out of 45 patients (36 %). It may be concluded that HER-2/neu is overexpressed in a substantial minority of CUP cases.
Massard et al. reported expression of EGF receptor (EGFR), which is important as a therapeutic target, in 66 % among 54 cases, while the proportion of c-KIT expression and HER-2/neu overexpression were 10 % and 4 % in this study, respectively [29]. Furthermore, the authors reported that EGFR expression was significantly correlated with response to cisplatin-based chemotherapy [29]. Dova et al. investigated 50 CUP cases and found EGFR expression in 74 % and strong expression in 12 % of cases [30]. From a clinical perspective, a more relevant finding would be activating EGFR mutations at the genetic level since they are regarded as predictor for response to anti-EGFR therapy, while immunohistochemical detection of EGFR expression is a poor predictor regarding this, although the absence of mutations does not preclude response to drugs targeting EGFR [31]. In this regard, the mentioned study by Dova et al. included genetic analyses of EGFR, but disappointingly, activating EGFR mutations were not found [30]. Another publication, which will be discussed in detail further below [32], reported EGFR positivity in 15 out of 69 cases (22 %). In conclusion, it seems that methodological differences impede comparisons between these studies and preclude any firm conclusions.
Another group of therapeutic targets are type III receptor tyrosine kinases including c-KIT (CD117) and the platelet-derived growth factor receptors PDGFRα and PDGFRβ, which can be targeted by imatinib and some of its derivatives [33]. As already mentioned, the percentage of cases positive for c-KIT reported by Massard et al. was only 10 % [29]. Dova and coworkers found c-KIT expression by immunohistochemistry in 30 out of 37 CUP specimens (81 %), 5 of which (13 %) exhibited strong positivity [34]. PDGFRα was positive in 15 (50 %) and strongly positive in 1 (3 %) out of 30 cases in this analysis [34]. As with EGFR, screening for mutations within exon 11 of c-KIT and exons 12 and 18 of PDGFRα did not reveal any mutations [34]. Even less data exist regarding PDGFRβ, which was positive in 17 out of 69 cases (25 %) in one study [32]. We conclude that data regarding type III receptor tyrosine kinases in CUP are sparse. Nevertheless, it might be assumed that these kinases do not play a major role in this disease.
More than 50 % of human cancers carry mutations in the TP53 gene, which is therefore regarded as the most important tumor suppressor [35]. Regarding its role in CUP, an early study was published in 1993, which comprised a screening for TP53 mutations within the exons 5–9 (which are most frequently affected by TP53 mutations) in a small series of 15 CUP biopsy specimens and eight cell lines established from CUP samples [14]. With this approach, TP53 mutations were detected in 26 % of cases, a proportion lower than usually found in other solid tumors. In contrast, a later study of 47 CUP cases using immunohistochemistry revealed expression of TP53 and BCL-2 in 70 % and 65 % and overexpression of these proteins in 53 % and 40 % of tumors, respectively [36]. Since TP53 overexpression correlates well with p53 mutation, these data suggest a frequency of mutant TP53 in the magnitude of 50 %, which would be closer to the percentages reported for other cancers. This finding was independently confirmed by van de Wouw et al. [28], who reported TP53 overexpression by immunohistochemistry in 23 out of 48 cases (48 %). Puzzlingly, others [32] reported TP53 positivity only in 13 out of 69 specimens (19 %). Another surprising finding relates to the CUP subgroup located to head and neck lymph nodes: Although TP53 mutations are a frequent finding in other head and neck carcinomas, sequencing of exons 4–9 of TP53 in 23 specimens from head and neck lymph node CUP cases revealed no mutations at all [37]. Together, the small sample sizes and methodological differences of the above studies again make it difficult to draw firm conclusions also on the TP53 mutation frequency in CUP.
The publications cited up to now mainly relied on immunohistochemistry, and if molecular analyses were included, they were restricted to selected exons. In contrast, the advent of next-generation sequencing has enabled researchers to speed up the investigation of tumor-associated mutations by several orders of magnitude with the additional advantage that the findings are less dependent on or biased by educated guessing. Not unexpectedly, it took some years from the introduction of this methodological advance until the first results with CUP samples were published. Not until last year, Tothill et al. [16] examined tumors from 16 CUP cases by sequencing a large panel of 701 genes, which was supplemented by an array-based profiling of copy-number alterations. Twelve cases displayed mutations, deletions, or amplifications of genes for which targeted treatment approaches are available, including mutations of KRAS, PIK3CA, AKT1, and IDH1 and a homozygous deletion of BRCA1 [16]. The authors claimed that in five of these cases, there was strong clinical evidence supporting the efficacy of a drug based on the tumor genotype [16]. In addition, they argued that comparison of the mutation spectra of CUP cases with published mutation frequencies from different solid tumor entities – which they combined with clinical and immunohistochemical clues and the results of gene expression profiling – was useful for assignment of a likely site of origin [16]. With regard to pathogenetic mechanisms, it was stated that the majority of actionable mutations and copy-number alterations were identified in core mitogenic and cell growth pathways [16], but this may be biased by the fact that most of the available targeted drugs are designed to target these pathways.
Related posts:
 Diagnostic Work-up of CUP: An Approach to Current Recommendations and Underlying Evidence
Treatment of Disseminated CUP
Future Directions
Diagnostic Work-up of CUP: An Approach to Current Recommendations and Underlying Evidence
Treatment of Disseminated CUP
Future Directions
 Concepts of Radiotherapy in Cancer of Unknown Primary
Concepts of Radiotherapy in Cancer of Unknown Primary
 Favorable Subsets Among Cancers of Unknown Primary
Favorable Subsets Among Cancers of Unknown Primary
 Epidemiology, Risk Factors, and Survival in CUP: Pointers to Disease Mechanisms
Epidemiology, Risk Factors, and Survival in CUP: Pointers to Disease Mechanisms
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


