Biologic and Pharmacologic Basis of Cancer Chemotherapy
Biologic and Pharmacologic Basis of Cancer Chemotherapy
Roland T. Skeel
I. GENERAL MECHANISMS BY WHICH CHEMOTHERAPEUTIC AGENTS CONTROL CANCER
The purpose of treating cancer with chemotherapeutic agents, whether traditional or targeted, is to prevent cancer cells from multiplying, invading, metastasizing, and ultimately killing the patient. Most traditional chemotherapeutic agents appear to exert their effect primarily on cell proliferation. Because cell multiplication is a characteristic of many normal cells as well as cancer cells, most nontargeted cancer chemotherapeutic agents also have toxic effects on many normal cells, particularly those with a rapid rate of turnover, such as bone marrow and mucous membrane cells. The goal in selecting an effective drug in this category, therefore, is to find an agent that has a marked growth-inhibitory or controlling effect on the cancer cell and a minimal toxic effect on the host. In the most effective chemotherapeutic regimens, the drugs are capable not only of inhibiting but also of completely eradicating all neoplastic cells while sufficiently preserving normal marrow and other target organs to permit the patient to return to normal, or at least satisfactory, function, duration, and quality of life.
Inhibition of cell multiplication and tumor growth can take place at several levels within the cell and its environment:
Macromolecular synthesis and function
Cytoplasmic organization and signal transduction
Cell membrane and associated cell surface receptor synthesis, expression, and function
Environment of cancer cell growth
A. Classic chemotherapy agents
Most traditional chemotherapy agents (not including immunotherapeutic agents, other biologic response modifiers, and molecular targeted therapies) appear to have their primary effect on either macromolecular synthesis or its function.1 This effect means that they interfere with the synthesis of DNA, RNA, or proteins or with the appropriate functioning of the preformed molecule. When interference in macromolecular synthesis or function in the neoplastic cell population is sufficiently great, a proportion of the cells die. Some cells die because of the direct effect of the chemotherapeutic agent. In other instances, the chemotherapy may trigger differentiation, senescence, or apoptosis, the cell’s own mechanism of programmed death.
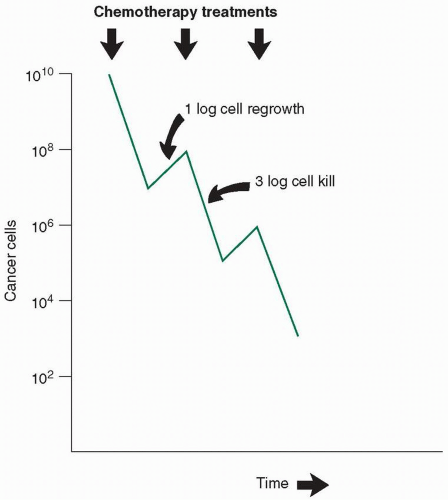
Cell death may or may not take place at the time of exposure to the drug. Often, a cell must undergo several divisions before the lethal event that took place earlier finally results in the death of the cell. Clinical responses may thus be delayed even when the treatment has effectively put the tumor cells into a death spiral. Because only a proportion of the cells die as a result of a given treatment, repeated doses of chemotherapy must be used to continue to reduce the cell number (Fig. 1.1). In an ideal system, each time the dose is repeated, the same proportion of cells—not the same absolute number—is killed. In the example shown in Figure 1.1, 99.9% (3 logs) of the cancer cells are killed with each treatment, and there is a 10-fold (1-log) growth between treatments, for a net reduction of 2 logs with each treatment. Starting at 1010 cells (about 10 g or 10 cm3 leukemia cells), it would take five treatments to reach fewer than 100, or 1, cell. Such a model makes certain assumptions that rarely are strictly true in clinical practice2,3:
FIGURE 1.1 The effect of chemotherapy on cancer cell numbers. In an ideal system, chemotherapy kills a constant proportion of the remaining cancer cells with each dose. Between doses, cell regrowth occurs. When therapy is successful, cell killing is greater than cell growth.
All cells in a tumor population are equally sensitive to a drug.
Drug accessibility and cell sensitivity are independent of the location of the cells within the host and of local host factors such as blood supply and surrounding fibrosis.
Cell sensitivity does not change during the course of therapy.
The lack of curability of most initially sensitive tumors is probably a reflection of the degree to which these assumptions do not hold true.
B. Biologic response modifiers and molecular targeted therapy
There are intricate interrelated mechanisms within individual cells and cell populations that promote or suppress cell proliferation, facilitate invasion or metastasis when the cell is malignant, lead to cell differentiation, promote (relative) cell immortality, or set the cell on the path to inevitable death (apoptosis). These activities are controlled in large part by normal genes and, in the case of cancer by mutated constitutive genes, cancer promoter genes, tumor suppressor genes, and their products. Included in these products are a host of cell growth factors that control the machinery of the cell. Some of these factors that affect normal cell growth have been biosynthesized and are now used to enhance the production of normal cells (e.g., epoetin-α and filgrastim) and to treat cancer (e.g., interferon).
The recent expansion of our understanding of the biologic control of normal cells and tumor growth at the molecular level has begun to offer improved therapy for many types of cancer, such as melanoma and kidney cancer that formerly were quite resistant to traditional chemotherapy, and has helped to explain differences in response among cancers, formerly thought to be similar such as diffuse large cell lymphoma. New discoveries in cancer cell biology have provided insights into apoptosis, cell cycling control, angiogenesis, metastasis, cell signal transduction, cell surface receptors, differentiation, and growth factor modulation. New drugs in clinical trials have been designed to block growth factor receptors, prevent oncogene activity, block the cell cycle, restore apoptosis, inhibit blockade of immune recognition and control, inhibit angiogenesis, restore lost function of tumor suppressor genes, and selectively kill tumors containing abnormal genes. Further understanding of each of these holds a great potential for providing powerful and more selective means to control neoplastic cell growth and have already led to more effective cancer treatments in the past decade.4 The fundamental principles related to this group of antineoplastic agents are discussed in Chapter 2.
II. TUMOR CELL KINETICS AND CHEMOTHERAPY5
Cancer cells, unlike other body cells, are characterized by a growth process whereby their sensitivity to normal controlling factors has been partially or completely lost. As a result of this uncontrolled growth, it was once thought that all cancer cells grew or multiplied faster than normal cells and that this growth rate was primarily responsible for the sensitivity of cancer cells to chemotherapy. Now it is known that most cancer cells grow less rapidly than the more active normal cells such as bone marrow. Thus, although the growth rate of many cancers may be faster than that of normal surrounding tissues, growth rate alone cannot explain the greater sensitivity of cancer cells to chemotherapy.
A. Tumor growth. The growth of a tumor depends on several interrelated factors.6
1. Cell cycle time or the average time for a cell that has just completed mitosis to grow, redivide, and again pass through mitosis determines the maximum growth rate of a tumor, but probably does not determine drug sensitivity. The relative proportion of cell cycle time taken up by the DNA synthesis phase may relate to the drug sensitivity of some types (synthesis phase-specific) of chemotherapeutic agents.
2. Growth fraction or the fraction of cells undergoing cell division contains the portion of cells that are sensitive to drugs whose major effect is exerted on cells that are dividing actively. If the growth fraction approaches 1 and the cell death rate is low, the tumor-doubling time approximates the cell cycle time.
3. Total number of cells in the population (determined at some arbitrary time at which the growth measurement is started) is clinically important because it is an index of how advanced the cancer is; it frequently correlates with normal organ dysfunction. As the total number of cells increases, so does the number of resistant cells, which in turn leads to decreased curability. Large tumors may also have greater compromise of blood supply and oxygenation, which can impair drug delivery to the tumor cells as well as impair sensitivity to both chemotherapy and radiotherapy.
4. Intrinsic cell death rate of tumors is difficult to measure in patients, but probably makes a major and positive contribution by slowing the growth rate of many solid tumors.
B. Cell cycle
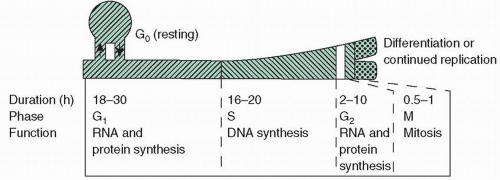
The cell cycle7 of cancer cells is qualitatively the same as that of normal cells (Fig. 1.2). Each cell begins its growth during a postmitotic period, a phase called G1, during which enzymes necessary for DNA production, other proteins, and RNA are produced. G1 is followed by a period of DNA synthesis (S phase), in which essentially all DNA synthesis for a given cycle takes place. When DNA synthesis is complete, the cell enters a premitotic period (G2), during which further protein and RNA synthesis occurs. This gap is followed immediately by mitosis, at the end of which actual physical division takes place, two daughter cells are formed, and each cell again enters G1. G1 phase is in equilibrium with a resting state called G0. Cells in G0 are relatively inactive with respect to macromolecular synthesis and are consequently insensitive to many traditional chemotherapeutic agents, particularly those that affect macromolecular synthesis.
FIGURE 1.2 Cell cycle time for human tissues has a wide range (16 to 260 hours), with marked differences among normal and tumor tissues. Normal marrow and gastrointestinal lining cells have cell cycle times of 24 to 48 hours. Representative durations and the kinetic or synthetic activity are indicated for each phase.
C. Phase and cell cycle specificity
Most classic chemotherapeutic agents can be grouped according to whether they depend on cells being in cycle (i.e., not in G0) or, if they depend on the cell being in cycle, whether their activity is greater when the cell is in a specific phase of the cycle. Most agents cannot be assigned to one category exclusively. Nonetheless, these classifications can be helpful for understanding drug activity.
1) Limitation to single-exposure cell kill. With a phase-specific agent, there is a limit to the number of cells that can be killed with a single instantaneous (or very short) drug exposure because only those cells in the sensitive phase are killed. A higher dose kills no more cells.
2) Increasing cell kill by prolonged exposure. To kill more cells requires either prolonged exposure to, or repeated doses of, the drug to allow more cells to enter the sensitive phase of the cycle. Theoretically, all cells could be killed if the blood level or, more importantly, the intracellular concentration of the drug remained sufficiently high while all cells in the target population passed through one complete cell cycle. This theory assumes that the drug does not prevent the passage of cells from one (insensitive) phase to another (sensitive) phase.
TABLE 1.1 Examples of Cell Cycle Phase-Specific Chemotherapeutic Agents
Phase of Greatest Activity
Class
Type
Characteristic Agents
Gap 1 (G1)
Natural product
Enzyme
Asparaginase
Hormone
Corticosteroid
Prednisone
G1/S junction
Antimetabolite
Purine analog
Cladribine
DNA synthesis
Antimetabolite
Pyrimidine analog
Cytarabine, fluorouracil, gemcitabine
Antimetabolite
Folic acid analog
Methotrexate
Antimetabolite
Purine analog
Thioguanine, fludarabine
Natural product
Topoisomerase I inhibitor
Topotecan
Miscellaneous
Substituted urea
Hydroxyurea
Gap 2 (G2)
Natural product
Antibiotic
Bleomycin
Natural product
Topoisomerase II inhibitor
Etoposide
Natural product
Microtubule polymerization and stabilization
Paclitaxel
Mitosis
Natural product
Mitotic inhibitor
Vinblastine, vincristine, vindesine, vinorelbine
Natural product analog of epothilone B
Mitotic inhibitor that binds to β-tubulin on microtubules
Ixabepilone
Only gold members can continue reading. Log In or Register to continue