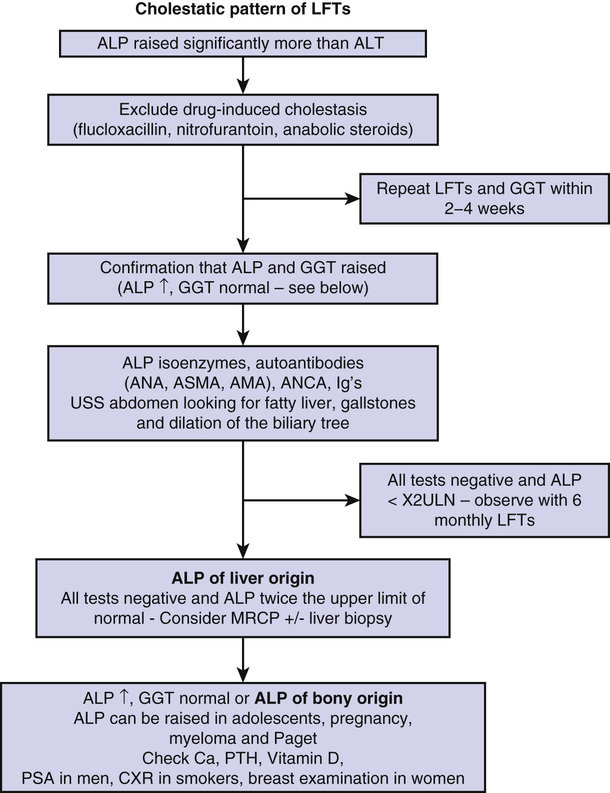
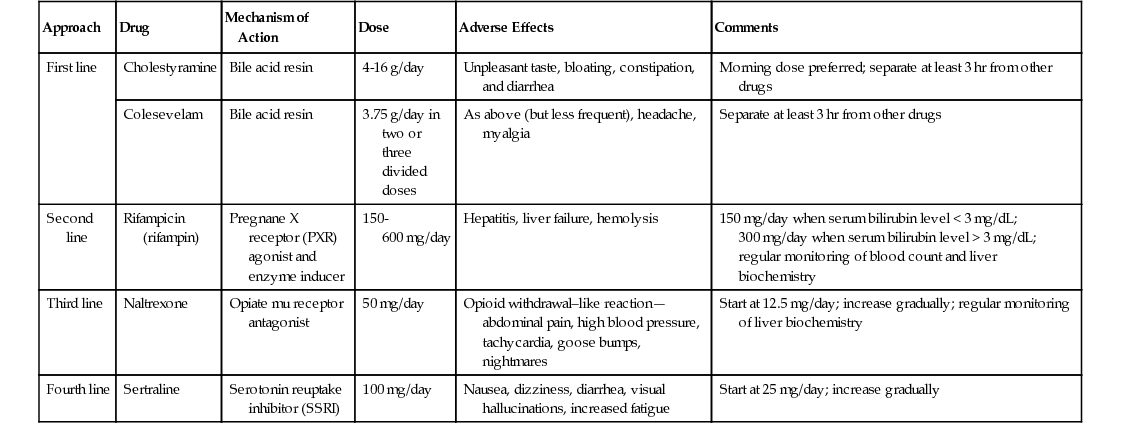
Noor Mohammed, Vinod S. Hegade, Sulleman Moreea Chapter 74 describes diseases of the liver as they affect older adults. This chapter will examine the related disorders of the biliary tract. Biliary tract disease is suspected when liver function tests (LFTs) are suggestive of cholestasis; the alkaline phosphatase (ALP) level is disproportionately higher than the ratio of the serum transaminase levels of alanine transaminase (ALT) and aspartate transaminase (AST; ALT/AST). A raised ALP level is often associated with a raised bilirubin level, and patients appear clinically jaundiced when the bilirubin concentration in the blood exceeds 51 µmol/L (3 mg/dL). Biliary tract diseases can be divided into intrahepatic and extrahepatic causes (Table 75-1). TABLE 75-1 Causes of Raised Alkaline Phosphatase Levels* ALP is coded by three separate genes and found in many locations in the body—liver, bone, placenta, kidneys, and intestine.1 Its precise function is unknown. In the liver it appears to downregulate the secretory activities of the intrahepatic biliary epithelium.2 Its half-life is 7 days, its site of degradation is unknown, and its clearance from serum is independent of the functional capacity of the liver or the patency of the bile ducts.3 The intestinal contribution (≈10% to 20%) is of limited clinical importance.4 Subjects older than 60 years have higher ALP values (up to 1.5 times normal) than younger adults, from the liver in older men and from bone in postmenopausal women.5 If there is an isolated increase in ALP, it is important to ascertain that this is of liver origin. For practical purposes in older adults, we mainly need to establish whether the raised ALP level is of liver or bone origin. The most practical approach is to measure the γ-glutamyl transpeptidase (GGT) level, whose elevation may also reflect biliary disease. However, GGT is an inducible enzyme that is not completely specific for hepatobiliary disease. It is elevated in people who drink large quantities of alcohol6 or take medicines such as phenytoin.7 ALP isoenzyme levels can be requested with the caveat that the test is heat-labile; bone and liver isoenzymes differ only slightly in electrophoretic mobility, sometimes leading to inconclusive results. 5′-Nucleotidase is a more specific biliary enzyme than GGT but is not measured routinely.8 Refer to the investigative pathway for raised ALP levels shown in Figure 75-1. When a patient is jaundiced with cholestatic LFTs, a thorough history is essential. The key points in the history are duration of jaundice and whether it is associated with abdominal pain, anorexia, weight loss, alcohol intake, and any past medical history of note. The examination is geared to the signs of chronic liver disease, evidence of weight loss, abdominal tenderness, and presence of any mass or organomegaly. Investigations should include a noninvasive liver screen (liver autoantibodies, immunoglobulins, AFP), and an urgent ultrasound (US) examination is requested. If the US scan shows biliary dilation with an obvious cause, further investigation and management will follow accordingly. If there is biliary dilation without any obvious cause, magnetic resonance cholangiopancreatography (MRCP) is requested, proceeding to an endoscopic US scan if necessary. If there is no biliary dilation, intrahepatic causes of cholestasis are the next targets of further investigations; see Table 75-1. An isolated raised bilirubin level may be seen in older patients and is not an indication of biliary tract disease. It is due to an increase in bilirubin production (e.g., via hemolysis, ineffective erythropoiesis, blood transfusion, resorption of hematomas) or decreased hepatocellular uptake or conjugation (e.g., congenital hyperbilirubinemias). Liver function is otherwise normal, and hyperbilirubinemia is often characterized by a predominant elevation in unconjugated bilirubin levels. Jaundice resulting from hemolysis is usually mild, with a serum bilirubin level of 68 to 102 µmol/L (4 to 6 mg/dL) because normal liver function can easily handle the increased bilirubin derived from excessive breakdown of red blood cells. Unconjugated bilirubin is not water- soluble and therefore will not pass into the urine—hence, the term acholuric jaundice. The urinary urobilinogen level is increased. Causes of hemolytic jaundice are the same as those of hemolytic anemia. Investigations have shown a raised unconjugated bilirubin level but normal serum alkaline phosphatase, transferase, and albumin levels, and serum haptoglobin levels are low. Most of the congenital and inherited defects are diagnosed in young people; the only condition that could incidentally be found in older patients is Gilbert syndrome, the most common familial hyperbilirubinemia. It is asymptomatic and is usually detected as an incidental finding of a slightly raised bilirubin level, 17 to 102 µmol/L (1 to 6 mg/dL) on a routine check. No signs of liver disease are present. There is a family history of jaundice in 5% to 15% of patients. Hepatic glucuronidation is approximately 30% of normal, resulting in an increased proportion of bilirubin monoglucuronide in bile. Most patients have reduced levels of uridine 5′-diphosphoglucuronosyltransferase (UDP) glucuronosyl transferase activity, the enzyme that conjugates bilirubin with glucuronic acid. The major importance of establishing this diagnosis is to inform the patient that it is not a serious disease and prevent unnecessary investigations. Tests show only a raised unconjugated bilirubin level, which is further increased when fasting, during mild illnesses or infections, after surgery, or during consumption of large amounts of alcohol; the reticulocyte count is normal. No treatment is necessary. Magnetic resonance imaging (MRI) is now the investigation of choice for characterization and staging of hepatic, pancreatic and splenic lesions and for imaging the biliary tree using MRCP. It does not involve the use of radiation. MRCP is commonly used for the diagnosis of gallstones, assessment of common bile ducts, intrahepatic ducts, or pancreatic ducts for stenotic lesions or stones, and investigating pancreatitis or abdominal pain of uncertain cause. A strong magnetic field is used to align rotating hydrogen protons within the tissue being imaged, and T1 and T2 relaxation times are used to create the final image. In T2-weighted images, water appears white and the liver dark, which makes the biliary tree easy to visualize, hence making MRCP the investigation of choice for biliary pathology. Older patients can find MRI difficult to tolerate because they have to lie supine and still for at least 30 minutes. Noise levels can be high because of vibration of the magnetic coils, and ear plugs or headphones need to be worn. Claustrophobia due to the tight cylindrical scanner can lead to a significant number of patients being unable to complete the test. Sedation can be used to improve compliance. Open coil magnets are increasingly being used for claustrophobic patients and for patients who are too large for the cylindric scanner. MRI contrast agents (e.g., gadolinium) are safer than the iodinated contrast agents used for CT scanning and rarely cause contrast reactions. However, they can cause contrast nephropathy and, in rare cases, nephrogenic systemic fibrosis in patients with moderate to severe chronic kidney disease. Contraindications include implanted devices such as cardiac pacemakers, implantable cardioverter-defibrillators (ICDs), nerve stimulators, cochlear implants, and embedded metallic foreign bodies such as intraorbital metallic fragments that may have been present from the working days of the now-retired patients. Transdermal patches need to be removed prior to MRI. There is a risk of contrast nephropathy with gadolinium-based contrast agents in those with advanced chronic kidney disease. The following are not contraindications and are considered safe for MRI: joint prostheses, coronary or peripheral vascular stents, prosthetic heart valves, sternal wires, inferior vena cava filters, and embolization coils. The composition of intracranial aneurysmal clips needs to be ascertained because most may not be safe during MRI. Primary biliary cirrhosis (PBC) is an autoimmune cholestatic liver disease of unknown cause, characterized biochemically by a cholestatic pattern of abnormal LFTs, serologically by the presence of antimitochondrial antibody (AMA), and pathologically by apoptotic damage to the biliary epithelial cells lining the small intrahepatic ducts. PBC predominantly affects middle-aged women, with a female-to-male ratio of 10 : 1 and the median age of disease onset about 50 years.9 The prevalence of PBC has a considerable geographic and regional variation, ranging from 128 to 180/million in Sweden10,11 to 150 to 400/million in the United States.12,13 In the United Kingdom, it ranges from 200 to 250/million, with regional differences.14,15 The pathogenesis of PBC is characterized by immune cell activation and damage to cholangiocytes, producing intrahepatic bile duct damage. The resulting cholestasis causes direct hepatocyte injury, leading to inflammation, necrosis, and ultimately resulting in liver fibrosis and cirrhosis. PBC should be suspected in the setting of chronic cholestasis after the exclusion of other causes of liver diseases. However, in older patients, an isolated rise in ALP levels needs further evaluation to rule out other conditions, such as Paget disease. The diagnosis of PBC is conventionally made using the combination of abnormal LFTs (elevation of serum ALP levels of liver origin for at least 6 months), the presence of AMA or PBC-specific serum antinuclear antibodies (ANAs), and/or a liver biopsy. AMAs are the characteristic serologic hallmark of PBC, and a titer of more than 1 : 40 in the context of cholestatic liver biochemistry is over 95% sensitive and specific for the diagnosis.16,17 Most patients with PBC have mild elevations of aminotransferase levels (ALT or AST) and increased levels of immunoglobulins (mainly IgM). Serum cholesterol levels are often elevated in PBC patients. Liver biopsy is not routinely indicated in the diagnosis of PBC nor is it essential before initiating treatment with ursodeoxycholic acid (UDCA). However, it can further substantiate the diagnosis and stage the liver disease. Histologically, PBC is characterized by chronic nonsuppurative cholangitis affecting small intrahepatic bile ducts, mainly interlobular and septal ducts. Focal duct obliteration with granuloma formation has been termed a florid duct lesion and, when present, it is considered as pathognomonic of PBC.18 In later stages of the disease, inflammation is seen in the hepatic parenchyma, with septal or bridging fibrosis eventually leading to cirrhosis, with regenerative nodules.19 Genome-wide association studies (GWAS) and iCHIP studies of PBC have been undertaken in those of European or Japanese ancestry and have shown that the human leukocyte antigen (HLA) complex makes an important contribution to the genetic basis of PBC. Risk haplotypes associated with PBC include those carrying DRB1*08 and DRB1*04 alleles. Protective haplotypes include those carrying DRB1*11 and DRB1*15 alleles. A total of 27 non-HLA risk loci for PBC have also been identified, harboring highly plausible candidate genes that are mainly involved in innate or acquired immune processes.20 Although most PBC patients are asymptomatic at the time of diagnosis, two characteristic symptoms of the disease, fatigue and pruritus (itch), can occur at any point in the disease course. Although the prevalence of fatigue (up to 78% of patients) and pruritus (20% to 70%) are similar,21 pruritus is a more specific symptom of PBC than fatigue. Both fatigue and pruritus are complex symptoms, and their cause in PBC remains incompletely understood. Also, for reasons that are currently unknown, neither fatigue nor pruritus correlates with the severity, histologic stage, or duration of PBC. Fatigue is often constant over time but pruritus may diminish in severity, especially during late stages of diseases. A recent study has observed that the intensity of itch depends on the age at disease presentation. In this study, younger patients (<30 years) scored 64% higher in pruritus score than those who presented after 70 years of age.22 Nocturnal pruritus will affect sleep, worsen fatigue, cause mood changes and cognitive impairment, and impair the overall quality of life. Other features of PBC include dry eyes and/or dry mouth (sicca syndrome), xanthelasma, xanthomata, portal hypertension, osteoporosis, and hyperlipidemia. UDCA is the only drug licensed by the U.S. Food and Drug Administration (FDA) for the treatment of PBC. UDCA is a bile acid that acts as a choleretic agent, with fewer hepatotoxic properties than endogenous bile acids.19 In a number of prospective randomized controlled trials, its use at more than 10 mg/kg of body weight (BW)/day has been shown to improve liver biochemistry significantly, slow histologic progression (in those with early stage disease), reduce the risk of developing varices, and improve liver transplant–free survival of patients.23 However, it has not been shown to be effective in reducing pruritus or improve fatigue. Current guidelines recommend the use of UDCA for patients with PBC who have positive tests for AMAs and elevated liver biochemical markers. The optimal dose of UDCA is 13 to 15 mg/kg BW/day, and it is be used for patients at any stage of PBC, as long as the liver biochemistry is abnormal.21 UDCA is generally well tolerated, with occasional side effects of diarrhea, abdominal discomfort, nausea, and vomiting. The benefit of UDCA for patients with positive AMA tests and normal liver biochemistry is less clear; therefore, these patients should not be considered for UDCA treatment. It is important to exclude other systemic and dermatologic causes of pruritus, and patients with skin lesions should be referred to a dermatologist. Worsening of pruritus has been reported with the use of UDCA. In such an event, a trial off UDCA should be attempted, with reintroduction of UDCA at a lower dose. Patients with mild localized pruritus should be offered topical treatment with aqueous cream and 1% menthol (emollient and coolant effect). For those with generalized itch, guidelines recommend four specific drugs: cholestyramine (first line) or colesevelam, rifampicin (second line), naltrexone (third line), and sertraline (fourth line).21 Table 75-2 provides a summary of these drugs. It is a common practice to use each drug as monotherapy for at least 4 weeks (in the absence of side effects) then switching to next-line therapy if no improvement in pruritus occurs. When taking cholestyramine or colesevelam, patients should be advised to separate taking UDCA and any other drugs by at least 3 hours to avoid interference with their absorption. In patients who benefit from cholestyramine but cannot tolerate its side effects, colesevelam should be offered because it has fewer side effects and may have better compliance. In patients with severe, medically refractory pruritus, invasive therapies that have been shown to be effective in case series are narrow-band ultraviolet B (UVB) phototherapy, endoscopic nasobiliary drainage (NBD), plasmapheresis, and molecular adsorbent recirculating system (MARS). Although these interventions are effective, they are not universally available, and the duration of relief from pruritus induced by these invasive therapies is variable, with a need for repeated sessions of treatments to maintain remission of pruritus. Currently, two large multicenter randomized clinical trials have been evaluating the role of ileal bile acid transport (IBAT) inhibitor in treating pruritus in PBC patients (ClinicalTrials.gov identifiers—NCT01899703 and NCT01904058). Fatigue is multifactorial and not a specific symptom of PBC, and other causes of fatigue such as anemia, hypothyroidism, depression, sleep disorder, and vitamin D deficiency should be considered and, when detected, appropriately treated.21 Currently, there is no effective therapy available for the treatment of fatigue in PBC. Randomized controlled trials have failed to show any benefit with ondansetron (antagonist of serotonin 3 receptor) and fluoxetine (serotonin reuptake inhibitor). An association between fatigue and excessive daytime sleepiness has been shown in patients with PBC.24 The use of modafinil (a central nervous system stimulant) at doses of 100 to 200 mg/day has been shown to be effective in significantly improving the fatigue in PBC patients with fatigue and daytime somnolence.25 A staged exercise program and antioxidant vitamins (e.g., ubiquinone, 100 mg/day) have also been suggested to relieve fatigue in PBC. Liver transplantation is the definitive treatment and has proven survival benefit for PBC patients with advanced-stage disease. Criteria for the recommendation of liver transplantation for a PBC patient are similar to those for other types of end-stage liver diseases. These include decompensated cirrhosis with an unacceptable quality of life or anticipated death within 1 year due to treatment-resistant ascites and spontaneous bacterial peritonitis, recurrent variceal bleeding, encephalopathy, and hepatocellular carcinoma.18 The post–liver transplantation outcome is favorable, with survival rates above 90% and 80% to 85% at 1 and 5 years, respectively. Primary sclerosing cholangitis (PSC) is a chronic cholestatic disease of unknown cause characterized by inflammation and progressive obliterating fibrosis of the intrahepatic or extrahepatic biliary ducts. It is a slowly progressive disease, eventually leading to biliary cirrhosis and liver failure and complicated by related malignancies, such as cholangiocarcinoma and colorectal cancers. The term primary is used to distinguish it from secondary conditions from bile duct strictures causing cholestasis. It is rare (8 to 13 cases/100,000 persons) and affects primarily white men during their fourth or fifth decade of life.26 However, a Japanese study has suggested a second peak in the seventh decade of life, with a bimodal age distribution.27 Immune (genetic) and nonimmune (e.g., infections, toxins, ischemic damage, nonsmoking) mechanisms have been postulated as possible causes of PSC. There is a strong association between PSC and inflammatory bowel disease (IBD), which has led to the identification of many shared genetic loci. Coexisting IBD is seen in 75% to 90% of PSC patients, ulcerative colitis (UC) is seen in around 87%, and Crohn disease (CD) in 13%.28 However, there is no correlation between the severity of PSC and IBD, nor is there a temporal relationship between the onset of the diseases. Furthermore, therapy of IBD has little effect on the course of PSC, and vice versa. Approximately 5% to 30% of patients develop cholangiocarcinoma, with the highest incidence being in the first year of diagnosis; the 5-year survival is only 20% to 40% for resectable disease.29,30 Individualized treatment for cholangiocarcinoma with liver transplantation and aggressive neoadjuvant chemotherapy has shown promising results. However, the International Liver Cancer Association does not recommend liver transplantation for those with intrahepatic cholangiocarcinoma.31 Patients may be asymptomatic and diagnosed accidentally on routine blood tests. Asymptomatic PSC is found in 20% to 40% of patients investigated for abnormal LFTs. Occasionally, the symptoms have a relapsing and remitting course. Typical symptoms on presentation are jaundice, right upper quadrant pain, pruritus, and fatigue. One third of patients have episodes of acute cholangitis, with recurrent attacks. Liver enzyme levels, and especially ALP levels, are raised in most patients; perinuclear antineutrophil cytoplasmic antibodies (pANCAs) may be detected in 26% to 85% of PSC cases, but they lack specificity for diagnosis. Overlap syndromes with other forms of liver disease have been described. PSC with autoimmune hepatitis-like features has been referred to as autoimmune cholangitis. These patients usually present with high serum ALT levels, modest or no elevations in serum alkaline phosphatase levels, high titers of ANAs and anti–smooth muscle antibodies (SMAs), and histologic liver findings typical of autoimmune hepatitis. Characteristic stricturing of biliary tracts is seen on MRCP. Dominant strictures develop during the disease course in about 36% to 63% of patients.32 This leads to further elevation of liver enzyme levels, usually ALP. Dominant strictures develop de novo or are due to diffuse inflammatory periductal fibrosis; more recently, Candida has been hypothesized as the causative infective organism.33,34 These are treated with endoscopic retrograde cholangiopancreatography (ERCP) and biliary stent insertions; however, the development of a dominant stricture is associated with poor outcome.35 Liver biopsy is rarely required to diagnose PSC. It shows periductal inflammation in the early stages, which then extends to periportal areas, leading to septal fibrosis and necrosis, eventually resulting in frank cirrhosis of the liver.36
Biliary Tract Diseases
General Considerations
Intrahepatic Causes
Extrahepatic Causes
Primary biliary cirrhosis (PBC)
Luminal causes—common bile duct stones (choledocholithiasis)
Primary sclerosing cholangitis (PSC)
Mural causes—benign and malignant biliary strictures
IgG4-related sclerosing cholangitis
Extramural causes (e.g., malignant obstruction)
Drug-induced liver injury (DILI) causing cholestasis
Infiltrative liver diseases (e.g., fatty liver, granulomatous liver diseases, metastatic malignancy, amyloidosis)†
Viral hepatitis (e.g. hepatitis E)†
Congestive cardiac failure†
Infectious hepatobiliary diseases in AIDS (e.g., tuberculosis, cytomegalovirus, microsporidiosis)†
Biliopathy following liver transplantation†
Liver Function Tests
Alkaline Phosphatase
Investigating Cholestatic Jaundice
Magnetic Resonance Imaging
Primary Biliary Cirrhosis
Genetics of Primary Biliary Cirrhosis
Clinical Features
Management
Treatment of Pruritus
Treatment of Fatigue
Liver Transplantation
Primary Sclerosing Cholangitis
Clinical Features
Investigations
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Biliary Tract Diseases
75