Fig. 3.1
Oncogenesis: Aberration accumulation and genomic complexity
3.2.1 Single-Nucleotide Variation (SNV)
These subtle changes of the genetic sequence, observed at the nucleic level, involve the modification, the deletion, or the insertion of one or few base pairs in the reference sequence. These mutations can appear during the DNA replication or can be caused by exogenous (X-rays, UV, carcinogenic chemical substances, etc.) or endogenous (free radicals produced during metabolism, etc.) agents.
From a chemical point of view, there are two types of point mutations: transition mutations and transversion mutations. Transition mutations occur when a pyrimidine base (i.e., thymine [T] or cytosine [C]) substitutes for another pyrimidine base or when a purine base (i.e., adenine [A] or guanine [G]) substitutes for another purine base, and transversions are a crossing exchange, i.e., a pyrimidine versus a purine and vice versa.
Biologically, three classes of point mutations can be described in addition to small insertions deletions:
When the indel is a multiple of three, the consequences are quite simple: a new codon is added or lost, and the neighboring one can also be modified, and even if the sequence is still in frame, it can affect the resulting protein. For example, EGFR exon 19 insertions are a family of EGFR-tyrosine kinase inhibitors (TKI)-sensitizing mutations, and patients with tumors harboring these mutations should be treated with EGFR-TKI (He et al. 2012; Kris et al. 2012).
Silent mutations
A silent mutation also called synonymous mutation addresses the same amino acid and has no functional effect on the protein. As 64 codons code only 20 amino acids (degeneracy of the genetic code), a single nucleotide can change, but the new codon specifies the same amino acid.
Missense mutations
A missense mutation changes a codon in a nonsynonymous manner so that a different amino acid is created. Functionally, these missense point mutations can be conservative or not. For conservative mutations, the properties of the modified amino acid remain the same (hydrophobic, hydrophilic, etc.) whereas for nonconservative ones, the resulting amino acid can confer different properties to the protein. Prediction tools based on physicochemical score assessment usually classify them as “not deleterious” or “tolerant” as they do not seem to have the potential to affect wild-type protein function. The protein may be activated (see the “gain-of-function” section), as illustrated with the BRAF p.V600E. This missense mutation changes a valine (codon 600) to glutamic acid and leads to an activation of the BRAF protein which causes an unlimited proliferative signaling in cancer cells. Dedicated theranostic approaches can therefore be applied with the use of vemurafenib treatment in mutant V600E-BRAF melanoma (Davies et al. 2002; Bollag et al. 2010; Chapman et al. 2011).
Nonsense mutations
A nonsense mutation converts an amino acid codon into a stop codon (TAA, TAG, TGA), which truncates the protein and impacts its function depending on the position of the stop codon in the frame of the protein.
Indels and in-frame or frameshift mutations
If the term SNV for single-nucleotide variation is commonly used to describe substitution, it can also be applied to insertions or deletions (indel) of a single or few base pairs.
However, as gene expression is based on the triplet nature of the codons when the indel affects a number of nucleotides that is not divisible by three, the insertion or deletion can change the reading frame, resulting in a completely different translation. The earlier in the sequence the deletion or insertion occurs, the more altered is the protein. The polypeptide being created could be abnormally short (prematured stop codon) or more rarely abnormally long and will most likely be non functional.
3.2.2 Chromosomal Alterations
The study of malignant cells karyotypes showed that the majority of the human cancers had lost and/or won some chromosomal materials also called chromosomal aneuploidy. These losses and gains, often generated by the same kind of mechanisms, can affect all or part of a chromosome, giving rise, respectively, to either numerical or structural chromosomal aberrations. While oncogenic potential of certain aberrations, like translocations or specific amplification, has been demonstrated to be strongly associated with specific cancers and contributes to their formation, other aberrations like large gains or losses appear to be less specific and random, even if in some cancers certain patterns of chromosomal aberrations can be recognized. The understanding of chromosomal aneuploidy and its mechanisms can help improve the knowledge of tumorigenesis (Albertson et al. 2006) (Fig. 3.2).
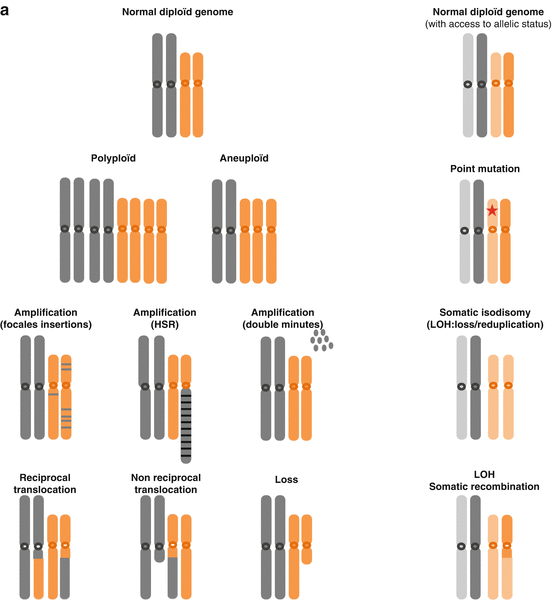
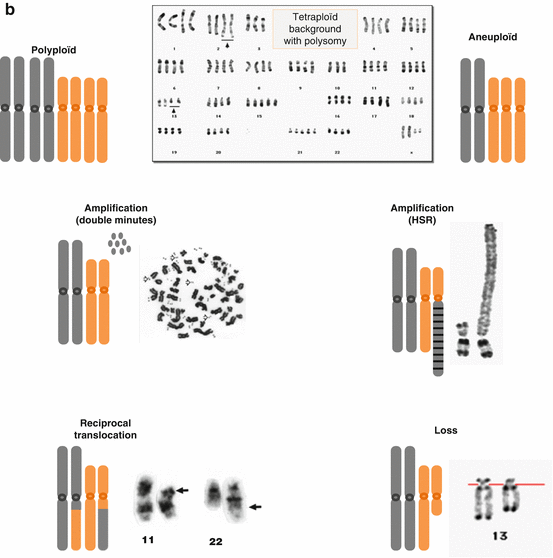
Fig. 3.2
(a) Examples of chromosomal alterations, theoretical point of view. (b) Examples of chromosomal alterations, chromosomal point of view (Adapted from Albertson et al. 2003)
3.2.2.1 Gains and Losses
Chromosome gains and losses are broad alterations encompassing from few megabases (Mb) to hundred Mb or even whole chromosomes. As these duplications (3–8 copies) or simple deletions (of a unique copy, if the tumor is diploid) are quite large, they can affect hundred of genes and their significance to the carcinogenesis process is less understood. Although there is evidence suggesting chromosome selection preferences for aneuploid cells in certain cancer types and although changes in gene copy number are among the most frequent molecular events in human cancer genome, these kinds of aberrations do not seem to be targetable.
3.2.2.2 Focal Amplifications
Certain tumors present genomic amplifications reflecting the acquisition of an increased number of DNA copies (from 8–10 to hundred sometimes) by multistep processes usually under positive selection. Often associated with late and/or aggressive stages of cancerous progress, these genomic inscreases can be infrachromosomal, and then called HSR for homogeneous staining region, or extrachromosomic as chromosomes double minute, which proposed mechanism is based on “bridge-fusion-break” iterations. Generally associated with the tumoral proliferation, the amplifications can be also found in initial phases in certain sarcomas. These addictions to a number of oncogenic drivers, leading to these amplifications, for example, genomically amplified MDM2 proteins, MET, HER2, and EGFR, are of particular interest because of pharmacological approaches which can directly target and inhibit the overexpressed protein (Fig. 3.3).
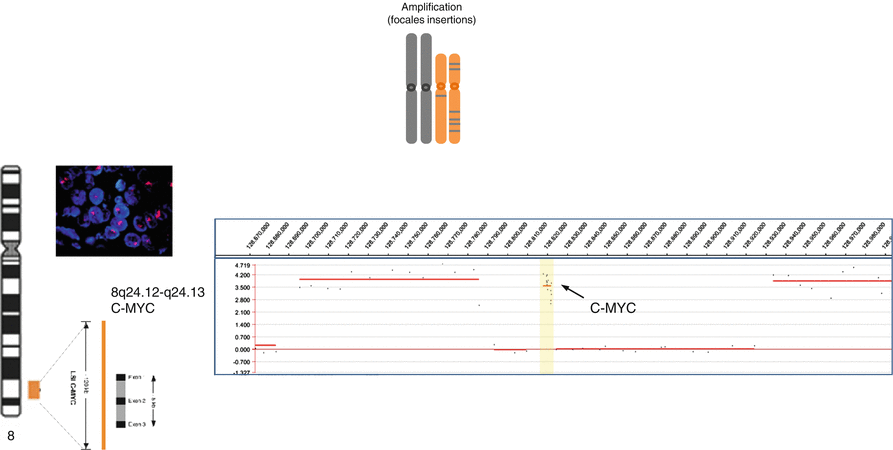
Fig. 3.3
Focal amplification (assessed by FISH and aCGH)
3.2.2.3 Homozygous Deletion
In a diploid organism, homozygous deletion refers to the loss of both alleles or a portion of both alleles from a genomic location. By extension, this physical loss affect both copies of the same gene or of the same chromosomal segment of a pair of homologous chromosomes. As multigenic deletion have severe consequences, and as the size of the deleted region is directly correlated to the number of the genes to be lost, the homozygous deletion is often small from few kilobases (Kb), which can arise within a tumor suppressor gene (intragenic deletion) to very few Mb affecting a small number of genes. Losses of PTEN, CDKN2A, RB1, or SMARCB1 are well-documented examples of genes inactivated by homozygous deletion. The double deleted region can be the intersection of two independent events, two large overlapping deletions (four distinct breakpoints), a small deletion associated to loss of the whole remaining homologous chromosome (only two different breakpoints on the remaining chromosome), and sometimes a small deletion with loss and reduplication of the chromosome also called somatic isodisomy.
3.2.2.4 Somatic Isodisomy
Isodisomy is a very specific type of genomic aberration in which the two copies of the chromosome originate from the same chromosome (inherited from one parent) with resultant homozygosity at all gene loci on the considered chromosome. Most often, isodisomy concerns an entire chromosome when the homologous chromosome is lost, but it may also be limited to part of a chromosome (segmental isodisomy). The presence of isodisomy in a tumor genome can highlight a simple mechanism to obtain biallelic inactivation of a tumor suppressor gene which is already mutated or disrupted on one allele (Fig. 3.4).
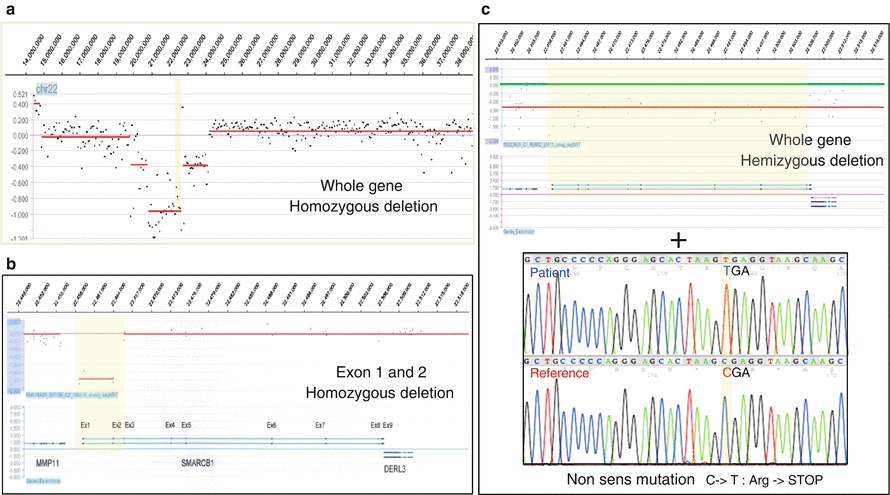
Fig. 3.4
Biallelic inactivation. (a) Whole-gene homozygous deletion (aCGH); (b) focal homozygous deletion within a gene (aCGH); (c) hemizygous deletion associated with nonsense point mutation (aCGH + Sanger sequencing)
3.2.2.5 Structural Aberration
As translocations directly affect a small number of genes, the role of many translocations in cancer causation has become much clearer over the years. These translocations can be within a chromosome (with the homologous chromosome or inversion inside the same chromosome). They can also occur between different chromosomes and in a nonreciprocal manner (often with loss of material) or can form ring chromosome.
3.2.2.6 Recurring Translocations
In 1960, studying chronic myeloid leukemia, Nowell and Hungerford observed the first cytogenetic reorganization acquired and recurring: the Philadelphia chromosome. This recombination between chromosomes 9 and 22 leads to the translocation t(9;22)(q34;q11) (Nowell and Hungerford 1960).
These chromosome translocations can have two types of consequences:
Upregulation of the expression of a gene. In this case, the reorganization juxtaposes the strong elements of regulation of the promoter of one a gene (like IgH or TCR) near an oncogene and increases its transcription (Ott et al. 2013).
The mechanism is particularly frequent in lymphoid malignancies, with the first studied example: the t(8;14)(q24;32) in Burkitt’s lymphoma. These translocations are mostly generated during V(D)J recombination.
Breakpoints formation of a fusion gene transcript appears to leukemia (CML) breakpoints on each chromosome occur in two genes leading to the formation of a fusion gene with new and oncogenic properties. The resulting fusion transcript appears to be highly specific like BCR-ABL in chronic myelogenous leukemia (CML).
The identification of fusion transcripts not only supports the diagnosis but provides the basis for the development of novel therapeutic strategies aimed at blocking the aberrant activity of chimeric proteins. Molecular biology and, in particular, cytogenetic and qualitative and quantitative RT-PCR technologies allow with high efficiency and specificity the determination of specific fusion transcripts resulting from chromosomal translocations. In prostate cancer, the TMPRSS2-ERG fusion is found in ∼50 % of all tumors, making it the single most common genetic lesion of this disease (Gopalan et al. 2009). In Ewing’s sarcoma (EWSR1-ETS) or alveolar rhabdomyosarcomas (PAX3/7-FOXO1), the fusion detection as specific marker is mandatory for molecular diagnosis.
3.2.2.7 Chromothripsis
The recent discovery of a new kind of massive chromosomal rearrangement in different cancers, named “chromothripsis” (chromo for chromosome, thripsis for shattering), has questioned the established models for a progressive development of tumors. Indeed, this phenomenon, which is characterized by the shattering of one or a few chromosome segments followed by a random reassembly of the fragments generated, occurs during one unique cellular event. Diverse situations can cause chromothripsis (radiations, telomere erosion, abortive apoptosis, etc.), and two express “repair routes” are used by the cell to chaotically reorganize the chromosomal regions concerned: nonhomologous end joining and repair by replicative stress (Korbel and Campbell 2013; Zhang et al. 2013)
3.2.3 Cancer Genes
Through the years, the definitions of the genes involved in cancers did not stop evolving. Historically, these were highlighted by their capacities to transform normal cells (discovery of c-Src via v-Src in Rous’s sarcoma) (Rous and Robertson 1917; Stehelin et al. 1976) or to revert a so-called “malignant” phenotype once introduced into cultured cells (experiments of somatic hybrids) (Ephrussi et al. 1969).
They were then characterized by the type and/or the number of mutation conferring them their tumoral abilities, but at the moment, their classification refers much more to their function and the modification of this one in the tumoral context. Thus, two big categories of genes involved in cancer can globally be defined: those whose alterations cause a gain of function and those whose aberrations lead to a loss of function.
3.2.3.1 “Gain-of-Function (GoF)” Genes
In their normal nonmutated state, GoF genes are called proto-oncogenes and are directly or indirectly involved in controlling the rate of cell growth and are implicated in the regulation of cell division. When altered by a genetic event, such as point mutation, translocation, or chromosome amplification, these cancer-promoting genes become then abnormally activated and named oncogenes.
Mutations affecting oncogenes are most often somatic, but germinal activating mutations have already been described (e.g. RET involved in multiple endocrine tumor type II and MET gene in hereditary papillary cancer of the kidney) (Santoro et al. 2002; Linehan et al. 2007).
The activation of GoF is dominant, the alteration of only one allele being sufficient to confer an oncogenic potential. The consequences of this activation can be seen at the expression level or at the DNA level of the targeted gene.
Examples of Gain-of-Function Mechanisms
The increase of the transcription of an oncogene can result from the translocation in its closeness of strong sequences of regulation (promoting sequences), which change the modulation of the native level of expression leading to an overexpression, like c-MYC in the t(8;14) translocation arising in Burkitt’s lymphoma. The mutation affecting the nucleotidic structure of an oncogene led to the synthesis of a functionally abnormal protein. So fusion genes resulting of chromosomes translocation can produce chimeric proteins such as BCR-ABL (Philadelphia chromosome marker) in chronic myeloid leukemia or truncated protein like NOTCH in lymphoblastic leukemia (Grabher et al. 2006). Lastly, point mutations can also drive the synthesis of proteins blocked in active configuration (like for K-RAS CTNNB1 (beta-catenin) (Eser et al. 2014) (Rosenbluh et al. 2014)), and focal oncogene amplifications are classical mechanisms of gain of function (like for CDK4 or MDM2).
Related posts:
 Assessment of Biomarkers’ Predictive Value of Efficacy
Assessment of Biomarkers’ Predictive Value of Efficacy
 Introduction: Rationale for Precision Medicine Clinical Trials
Introduction: Rationale for Precision Medicine Clinical Trials
 The Key Role of Pathology in the Context of Precision Medicine Trials
The Key Role of Pathology in the Context of Precision Medicine Trials
 Designs for Evaluating Precision Medicine Trials
Designs for Evaluating Precision Medicine Trials
 Microarrays-Based Molecular Profiling to Identify Genomic Alterations
Microarrays-Based Molecular Profiling to Identify Genomic Alterations
 Bioinformatics for Precision Medicine in Oncology
Bioinformatics for Precision Medicine in Oncology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


