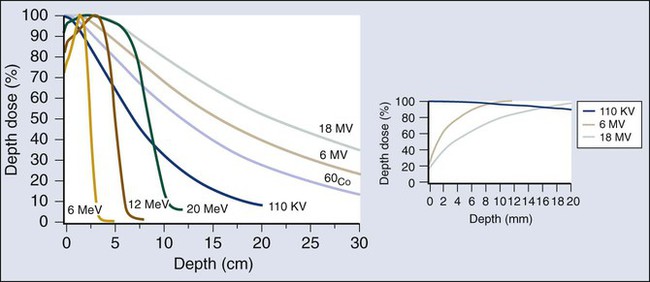
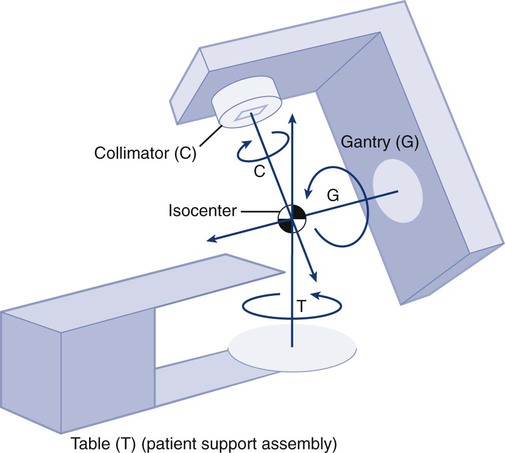
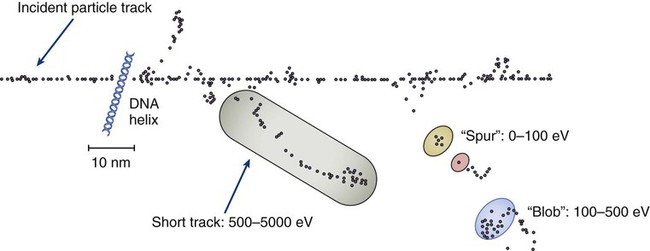
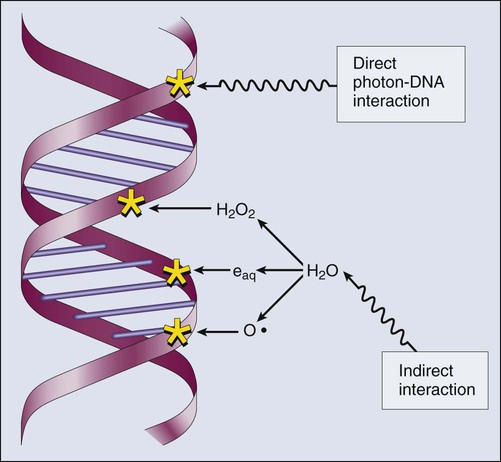
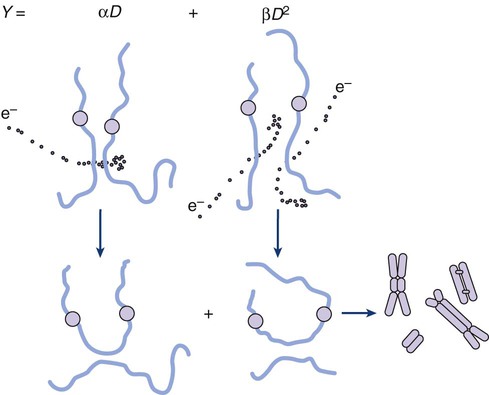
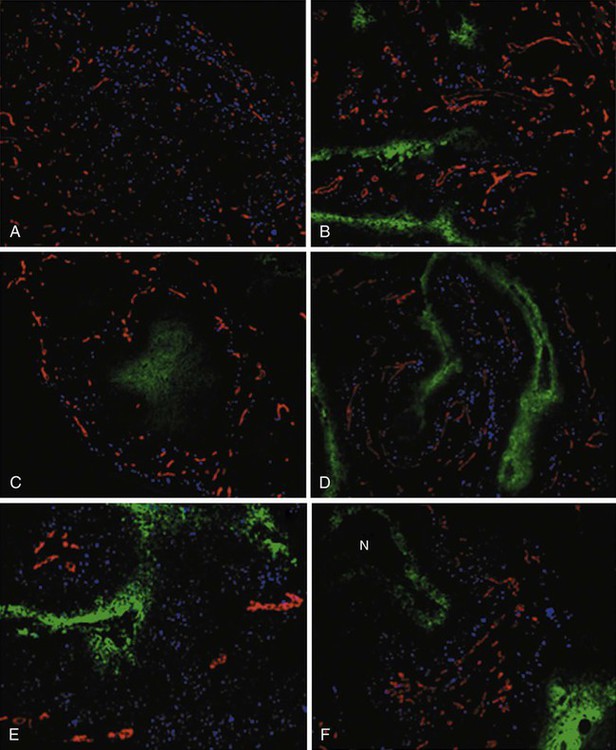
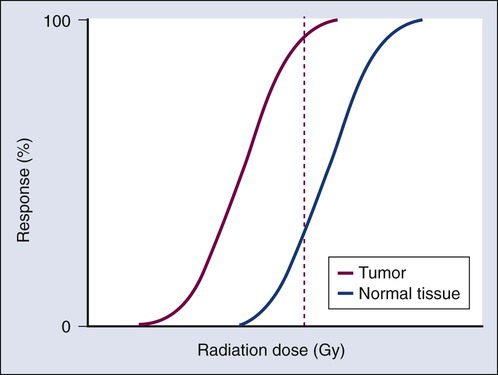
Elaine M. Zeman, Eric C. Schreiber and Joel E. Tepper Introduction and Historical Perspective • X-rays were first discovered emanating from an energized Crooke’s tube by Wilhelm Roentgen in 1895.1 In 1896, Henri Becquerel discovered that some naturally occurring elements emitted ionizing radiation.2 The radioactive elements radium and polonium were isolated and characterized by the Curies in 1898.3 • Within a year or two, ionizing radiation was in use worldwide for medical imaging and radiation therapy. • Several types of ionizing radiation are used to treat patients; most are of the low linear energy transfer, less biologically potent varieties. • Therapeutic x-rays (photons) and electrons are produced by linear accelerators but can also be produced by nuclear isotopes that undergo radioactive decay. These form the basis of external beam radiotherapy and brachytherapy, respectively. • Ionizing radiation interacts with matter via several processes, the most important of which for clinical radiation therapy is Compton scattering. • Megavoltage photons from linear accelerators have the desirable property of delivering their maximum dose at depth within the patient, thereby sparing the skin and, to some extent, other normal tissues. The Radiobiology of Radiotherapy • Ionization of biomolecules from the deposition of energy by photons or particles can occur directly and indirectly. The most important cellular target for radiation is DNA, with irreparable or “misrepaired” double-stranded breaks believed to be the lesions most responsible for cell killing. • Irradiation elicits diverse cellular responses that include the sensing of DNA damage, mobilization of DNA repair proteins, repair (or attempted repair) of DNA damage, triggering of cell cycle checkpoints, and, for irreparable or mis-rejoined damage, cell death by one of several mechanisms (e.g., mitotic catastrophe, apoptosis, and senescence). • The most commonly applied model of cell survival probability is the linear quadratic (α/β) model, with the surviving fraction of irradiated cells described by the equation • DNA damage and repair were initially inferred by monitoring increases in cell survival or tissue tolerance with fractionation. These phenomena were termed sublethal and potentially lethal damage repair or recovery. • Cells in different cell cycle phases possess different radiosensitivities; cells are most radiosensitive in the G2 and M phases of the cell cycle, and most resistant in the S phase, particularly the late S phase. Cells in the G1 phase are of intermediate radiosensitivity. • Well-oxygenated cells are as much as three times more sensitive to radiation-induced cell killing than (severely) oxygen-deprived cells. Viable hypoxic cells that exist in many human tumors but that are mostly absent in normal tissues may be an impediment to tumor control. The elimination of such cells has been a long-standing clinical goal. Hypoxia may provide avenues for therapeutic gain through the use of hypoxia-directed therapies. • Radiation sensitizers, particularly cytotoxic chemotherapy and, to a lesser extent, radiation protectors, aim to improve the therapeutic ratio. • Radiation therapy is used in more than half of all patients with cancer, either as an adjuvant or neoadjuvant treatment in combination with surgery; as a definitive treatment alone or in combination with chemotherapy; as an organ-sparing therapy; or to palliate symptoms. • Fractionation of radiation and altered fractionation schedules, such as accelerated hyperfractionated radiation therapy, make use of differences in the responses of normal and malignant tissues to irradiation to achieve higher therapeutic ratios. • Radiation produces early effects, such as mucositis, skin erythema, or desquamation, and late effects, such as fibrosis and carcinogenesis. Planning and Delivery of Radiation Treatment • Patient simulation uses multiple imaging approaches to identify cancerous and healthy regions within the patient and to select appropriate beams to deliver a dose to the tumor while minimizing the dose delivered to surrounding tissues. • Three-dimensional conformal treatment planning and delivery has permitted escalation of doses and improved sparing of normal tissues. • Intensity-modulated radiation therapy uses varying radiation beam intensities to precisely sculpt the dose distribution around the tumor to improve the therapeutic ratio. • Image-guided radiation therapy uses real-time and/or daily imaging to ensure that the tumor is positioned such that the radiation beams are precisely delivered to the appropriate location within the patient. • Brachytherapy delivers extremely high-dose radiation to tumor tissue with a much lower dose to surrounding normal tissues. • Stereotactic radiosurgery and stereotactic body radiation therapy combine a high dose per fraction with highly conformal treatment delivery to increase the therapeutic ratio while reducing treatment time. • Proton therapy has dose distribution advantages compared with photon therapy, and it may be used to deliver high doses of radiation to tumors in close proximity to sensitive normal structures. In order of roughly increasing energy, photons can interact with: 2. tightly bound inner shell electrons; 3. loosely bound outer shell electrons; 4. the extranuclear space surrounding the nucleus; or Photons having sufficient energy to ionize an atomic electron can undergo the photoelectric effect (Fig. 27-1). In this process, the photon energy is entirely absorbed. Some energy is lost to breaking the electron binding energy, and the rest is carried away as kinetic energy of the ejected electron. The probability of a photoelectric interaction scales with the cube of the atomic number (Z) and the inverse cube of the photon energy (E), making the photoelectric effect very sensitive to material type and much more prevalent for lower photon energies. The photoelectric effect is the dominant photon interaction in tissue below 30 keV. When photon energy is significantly higher than the binding energy of an electron, the photon can scatter from the electron without being absorbed, as illustrated in Figure 27-1. The result of this interaction is a photon with reduced energy and new direction and a recoil electron with some fraction of the initial photon energy. The energy of the scattered electron varies with the scattering direction. An electron scattered in the direction of the incident photon claims most of the initial photon energy, whereas electrons scattered at greater angles have successively less energy. Compton scattering is only weakly dependent on Z and is the dominant photon interaction in tissue between 30 keV and 30 MeV. Above 1.022 MeV, photons can interact in the presence of a strong nuclear field. The photon will disappear and spontaneously become an electron-positron pair (Fig. 27-1). The electron and positron will divide the initial photon energy between them to create their mass and kinetic energy. These particles will lose their energy as they interact with the surrounding materials. Upon losing all their energy, the electrons will be absorbed into an atom. The positron, on the other hand, will annihilate by interacting with a local electron, creating two 511 keV photons. (This annihilation reaction is what is detected during positron emission tomography scanning.) Pair production is the dominant atomic interaction in tissue for photons above 30 MeV and therefore has only a minor effect in radiation therapy, where energies are significantly lower. The most common modality used in radiation oncology is external beam radiation therapy. Although a small number of radiation therapy facilities generate external beams using radioactive sources such as cobalt-60, the vast majority of therapeutic electromagnetic radiation is generated in a linac. A linac is a device that accelerates charged particles (electrons) to velocities near the speed of light using oscillating electric fields to push the electrons through a series of accelerating cavities. A schematic of a linac is shown in Figure 27-2, A. Electrons are accelerated to energies typically between 4 and 18 MeV. Electric and magnetic fields focus and steer the high-energy electrons such that they strike a thin metal target that stops the electron beam, with some fraction of the electron energy converted to a spray of photons through the bremsstrahlung process. The bremsstrahlung photons, called x-rays, move approximately in the same direction as the electrons and have an energy spectrum, ranging from a few 10’s of keV up to the maximum energy of the initial electrons. The resulting photon beam then passes through a series of filters and beam-shaping elements that flatten and define the edges of the beam. The dose from a photon beam is related to its intensity, defined as the number of photons per unit area. Two major effects serve to decrease the intensity of a photon beam as it passes through tissue. First, as with any photon source, the beam intensity decreases with increasing distance from the source, just as is the case for a light bulb. In addition, beam intensity decreases as photons are attenuated from the beam via various scattering and absorption effects. This process leads to a characteristic decrease in intensity versus depth that varies based on photon energy. Although the photon intensity begins decreasing immediately upon entering a material, the energy released through the photon interactions is spread over a few centimeters as the electrons scattered by the photons gradually lose their energy as they pass through the material. The resulting dose distribution is characterized by a region of rapid increase near the surface, a leveling off of dose at a depth of 1 to 3 cm, and a gradual dose falloff as depth increases. The plot of dose versus depth is called a percent depth dose curve, as shown in Figure 27-3. Because higher energy photons are more penetrating, higher energy beams will attenuate more slowly, leading to a more gradual decrease in dose with depth. Linacs designed to produce photon beams can also be configured to produce therapeutic electron beams. Removing the photon-generating target and replacing it with a comparatively thinner electron scattering foil allows the transmission of the initial electron beam, but not without scattering the initially narrow beam into a broader distribution. Multiple filters and beam-shaping elements, as shown in Figure 27-2, B, produce an even distribution of customized shape at the surface of the patient. Electron beams lose their energy through different types of interactions than photons, leading to a different pattern of dose versus depth for electron beams. Rather than periodically removing photons from the beam through attenuation, electrons lose their energy gradually and at a relatively constant rate until the entire kinetic energy of the electron is expended and the particles simply stop. Ideally, this phenomenon would lead to a region of constant dose with increasing depth until the depth of full energy loss is reached, at which point the dose would drop abruptly to zero, with the depth of the dose drop-off being dependent on the initial electron energy. In practice, scattering and redirection of electrons within the beam lead to a mildly peaked dose plateau region and a somewhat more gradual dose falloff, as shown in Figure 27-3. Therapeutically useful isotopes vary in half-life and in energy of emitted particles, as shown in Table 27-1. Isotopes emitting higher energy particles can deliver significant amounts of dose farther from the radioactive source than those emitting lower energy particles. One type of radioactive source, 60Co, emits photons with an average energy of 1.25 MeV, which is sufficiently similar to photon energies found in linacs that 60Co can be used as an external source to treat targets deep in a patient. Most other therapeutically useful radioactive sources emit lower energy radiation with less penetrating power and must be placed in close proximity to the area to be treated. Sources are formed into small sealed seeds, typically 1 to 5 mm in size, and can be inserted into the treatment area on a temporary or permanent basis. The dose rate falls off very quickly with distance from a seed, both because of rapid attenuation and the rapid spread of photons as they move away from the source. Table 27-1 Therapeutically Useful Radioisotopes From Cox JD, Ang KK, editors. Radiation oncology: rationale, technique, results. 8th ed. St Louis: Mosby; 2003. The cytotoxic properties of ionizing radiation provide an opportunity for tumor control but also require that care be exercised to limit the exposure of healthy tissue to radiation. For external beam radiation therapy, linacs are typically mounted on rotating gantries (Fig. 27-4) that allow beams to pass through the patient and the target from a variety of directions. By placing the area to be treated at or near the center of rotation, multiple beams can be made to overlap in the region of the tumor, delivering a high dose to the overlap area and a comparatively low dose to other areas. For treatment based on implanted radioactive sources, a procedure known as brachytherapy, proper dose delivery consists of designing and delivering a three-dimensional (3D) distribution of radioactive seeds within the volume to be treated, creating a high-dose region that decreases rapidly beyond the treatment volume. Both of the aforementioned treatment modalities require a 3D understanding of the patient anatomy and require: The total amount of energy imparted to a cell during the passage of ionizing radiation (expressed in units of dose, Gray, which in turn is expressed in units of energy deposited, joules per kilogram) is by itself insufficient to describe its biological consequences. For example, 1 Gy of x-rays and 1 Gy of neutrons deliver the same total energy to a cell macroscopically speaking but do not produce equivalent biological effects because it is the microscopic pattern of that energy deposition, the spacing or density of the discrete ionization events along the track of the photon or particle, that is key to determining biological effectiveness. In this example, the 1 Gy of neutrons is much more potent biologically because the average energy deposited locally along the length of each neutron’s track is higher than for x-rays. This quantity of ionization density, typically expressed in units of keV/µm, is termed the radiation’s LET. The concept of ionization density is illustrated graphically in Figure 27-5 for radiations of differing LET, using a strand of DNA drawn to scale as a representative “biomolecule.” Although most radiation therapy is performed using low LET x-rays, γ-rays, or electrons, a few institutions do use high LET neutrons or even higher LET heavy charged particles (heavy ions), such as carbon ions, for treatment. Lower total doses of these radiations are used to achieve tumor control in keeping with their greater biological potency, and especially stringent limits are placed on the amount of normal tissue incidentally irradiated out of justifiable concern for an increased frequency of complications. The use of protons for radiotherapy is also gaining in popularity; protons behave like other high LET particles in terms of their physical properties, although somewhat surprisingly, they are only marginally more potent biologically than x-rays or electrons. Whether the ionization of a particular molecule results in a measurable biological effect depends on a number of factors, including how important the molecule is to the continued survival and function of the cell, how many copies of the molecule are normally present in the cell, and to what extent and how the cell responds to the loss of working copies. DNA is arguably the most important cellular macromolecule and one that is present only as a single, double-stranded copy, and thus an energy deposition event occurring directly in DNA certainly could affect a cell’s continued survival and functioning. Accordingly, much attention has been focused historically on understanding radiation-induced DNA damage and its repair and the consequences when that damage is either irreparable or “misrepaired.” That said, many other molecules in the cell may be less crucial to survival yet are much more abundant than DNA and therefore have a much higher probability of being ionized. By far, the most abundant molecule in the cell is water. Free radicals formed by the radiolysis of water (the hydroxyl radical, •OH, in particular) are capable of adding to the DNA damage resulting from direct energy absorption by migrating to the DNA and damaging it indirectly, as illustrated in Figure 27-6. This mechanism is referred to as “indirect radiation action” to distinguish it from “direct radiation action”4 previously described. Approximately 30% of the total DNA damage produced by a given dose of x-rays is from the direct effect, and 70% is from the indirect effect.4 Complex macromolecules like DNA that have been ionized and converted to free radicals undergo a series of chemical transmutations in an attempt to rid themselves of unpaired electrons, many of which involve the further breakage of chemical bonds. These broken bonds can result in the modification or loss of a DNA base or an entire nucleotide, a cross-linking of the two DNA strands, or a scission of the sugar phosphate backbone involving either one or both strands. Luckily, DNA is unique in that it is the only cellular macromolecule with its own repair system, and most of this damage can be repaired efficiently and typically with very high fidelity. Under certain circumstances, however, the cell’s attempts to repair these lesions may result in large segments of DNA being lost, rearranged, exchanged, or rejoined in inappropriate ways, so-called “misrepair.” In other cases, repair is impossible because of the complex nature of the damage itself, particularly when both DNA strands are involved (e.g., a DNA double-stranded break), or its location in the genome in time and space. It is this residual DNA damage that manifests itself as chromosome aberrations the next time the cell attempts to go through mitosis and that usually leads to cell death. In fact, the radiation dose response for the production of asymmetrical, exchange-type chromosome aberrations mirrors the shape of the corresponding cell survival curve.5 We now know that several different DNA repair pathways exist in mammalian cells, including single-step reactions that directly reverse certain simple types of damage, single and multistep base excision and resynthesis processes, and multistep pathways to “clean up,” resynthesize, and ligate single or double-stranded breaks in the DNA backbone.6 Which one (or more) of these repair pathways is activated depends on a number of factors, including the type of lesion produced, the physical location of the lesion in the genome (e.g., in the coding region of a gene versus in apparently noncoding DNA), the functional/temporal location of the lesion (i.e., in an actively replicating or transcribing gene versus an inactive one), and, critically, the overall repair competence of the cell in which the lesion was created.7 For a more in-depth discussion of the molecular biology, biochemistry, and regulation of DNA repair, please refer to Chapter 10. Many bacterial, yeast, rodent, and human genes involved in DNA repair processes have been identified and cloned,8 with many of the encoded proteins functioning as components of large repair complexes. Some of these proteins are interchangeable and participate in different DNA repair and replication pathways, whereas others are unique to specific types of repair. In fact, some are not directly involved with repair per se but rather link DNA repair to other cellular functions, including damage sensing, cell cycle checkpoint control, chromatin remodeling, and apoptosis.9 Inactivation or loss of any of these myriad proteins can lead to dysfunction of the DNA damage response, which in turn can precipitate diverse clinical syndromes of varying severity including immunodeficiency, neurodegenerative disorders, infertility, premature aging, hypersensitivity to DNA damaging agents, and cancer proneness.10 It is of particular interest that most cancer cells harbor one or more defects in the DNA damage response. Further, because different DNA repair pathways can share common components and functions under normal circumstances, and in some cases compensate for defects in other pathways, it may be that the clinical strategy of DNA repair inhibition will have a greater effect on the tumor than on normal tissues.10 The approach of combining DNA repair inhibitors with radiation to produce selective tumor radiosensitization is discussed further later in this chapter. The first activated oncogene identified to confer radiation resistance was NRAS (see references 11 and 12). Ras proteins, products of RAS family member genes, are small guanosine triphosphatases activated by upstream receptor tyrosine kinases. They transduce signals to a complex downstream cascade of protein kinases that have the net effect of promoting cell survival (for example, by the suppression of apoptotic cell death pathways) and unlimited proliferation (secondary to, for example, the downregulation of antiproliferative proteins), two of cancer’s hallmark phenotypes. When the ras protein is mutated, however, as is the case in approximately 30% of all human cancers, these pathways become overactive and no longer responsive to the normal regulatory mechanisms that antagonize the ras pathway and reign in excessive proliferation. This situation is exacerbated in many tumors by the concurrent loss of function of tumor suppressor proteins. Ras became, accordingly, a desirable target for molecular cancer drug development.13 The general approach to targeting ras focused on its need for posttranslational modification through prenylation to become activated.14 Despite encouraging preclinical results with a class of prenylation-inhibiting agents known as farnesyl transferase inhibitors—drugs that, when used alone or combined with radiation, produced sensitization of tumor cells with constitutively active ras pathways—these drugs did not meet expectations in clinical trials because of alternative prenylation pathways.13 Subsequent drug development focused on inhibiting proteins upstream of ras, in particular, members of the human epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases that transduce growth signals through ras and other signaling proteins.15,16 These transmembrane glycoproteins are activated through binding of ligands belonging to the EGF family of peptide growth factors. Cetuximab, a human-murine chimeric immunoglobulin G1 monoclonal antibody raised against the EGFR, is among the more successful molecularly targeted drugs, yielding improved outcomes in squamous cell head and neck cancers when combined with radiation therapy.17 Details of its mechanism of action and clinical use are discussed later in this chapter. Other classes of drugs that target EGFR include the small molecule tyrosine kinase inhibitors gefitinib and erlotinib, which inhibit EGFR phosphorylation and its downstream cascade by blocking the intracellular catalytic domain of the receptor.18 These drugs have shown some efficacy in selected patients with treatment-refractory, advanced non–small cell lung cancer (NSCLC).19 A second molecular target of interest is vascular endothelial growth factor (VEGF) and/or its cell surface receptor. VEGF is the most potent of the proangiogenic endothelial cell proliferation stimulators and chemoattractants and plays a pivotal role in promoting tumor survival by stimulating the growth of new blood vessels derived from the host vasculature. This phenotype of sustained angiogenesis is a hallmark property of cancer. Several isoforms of VEGF bind to the corresponding receptors VEGFR1, VEGFR2, and VEGFR3 (encoded, respectively, by the genes FLT1, KDR, and FLT4).20 Bevacizumab, a recombinant humanized monoclonal antibody, is the first molecularly targeted antiangiogenic drug to gain approval for clinical use by the U.S. Food and Drug Administration. This antibody binds to VEGF, with the net effect of eliminating signaling to vascular endothelial cells to initiate angiogenesis.21,22 The clinical use of bevacizumab in combination with radiation therapy is discussed later in this chapter. Death as a permanent, irreversible cessation of vital functions is not the same as what constitutes “death” to the radiation biologist or oncologist. For proliferating cells, including those maintained in vitro and the stem cells of both normal tissues and tumors in vivo, cell death in the radiobiological sense refers to a loss of reproductive integrity or clonogenicity, that is, an inability to sustain proliferation indefinitely. It is important to note that the term “clonogenic death,” as first described more than 50 years ago, is operationally defined and today serves as a catch-all term encompassing various mechanistic ways that cells die, all of which culminate in a cell losing its ability to divide indefinitely. These modes of cell death include mitotic catastrophe (the most common form of cell death after radiation exposure), apoptosis, autophagy, necrosis, senescence, and, strictly speaking, differentiation as well, to the extent that differentiated cells lose their ability to divide.23 Another noteworthy feature of clonogenic death is that it does not necessarily preclude the possibility that a cell may remain physically intact, metabolically active, continue its tissue-specific functions, and even divide a limited number of times after irradiation. The first report of a quantitative measure of intrinsic radiosensitivity for a human cell line (HeLa, derived from a cervical carcinoma) was published by Puck and Marcus in 1956.24 For different doses of x-rays, the reproductive integrity of HeLa cells was measured by their ability to form macroscopic colonies of at least 50 cells (corresponding to approximately six successful postirradiation cell divisions) on Petri dishes. The HeLa cell survival curve, in which the log of the surviving fraction of cells was plotted as a function of the radiation dose, was characterized by a roughly exponential dose response at intermediate to high doses and a bending, “shoulder” region at low doses where cell killing was less effective. This phenomenon is illustrated graphically in the upper panel of Figure 27-7. Radiation survival curves for hundreds of cell types derived from mammalian tumors and normal tissues have been generated in the years since the pioneering work of Puck and Marcus,24 and most are qualitatively similar to the original HeLa survival curve (see lower panel of Fig. 27-7). Mathematical models were developed to fit the cell survival data, with survival curve theory originating in a consideration of the physics of energy deposition in matter by ionizing radiation. An assumption inherent to target theory was that a biological response (cell killing in this case) resulted from critical “targets” receiving random “hits”25 in a probabilistic manner. Further, for cell survival curves with shoulders, each target was envisioned as requiring more than one hit to elicit the response, that is, that “sublethal” damage had to accumulate first before the cell would be killed. One mathematical expression derived from target theory that provided a fairly good fit to survival data was: Over time, it became apparent that some features of this model were inadequate,26 not the least of which was that its basis was the probabilistic nature of energy deposition in matter by ionizing radiation and not anything biologically based. For example, target theory was not concerned per se with which biomolecules in the cell were the purported “targets” of radiation damage, what the nature of the damage was in a molecular sense, or how the cell responded to it. (Of course, it was not lost on radiation biologists of the day that at least one cellular target was likely to be the DNA contained in chromosomes.24) A different and more biology-based interpretation of the dose response for radiation-induced cell killing was proposed by both Kellerer and Rossi27 and Chadwick and Leenhouts.28 The linear-quadratic (LQ) or “alpha-beta” equation, was shown to fit cell survival data quite well, particularly in the low-dose region of the curve where the target theory model often failed.26 In this expression, S is again the fractional cell survival following a dose (D), α is the rate of cell kill by a single-hit process, and β is the rate of cell kill by a two-hit mechanism. Implicit in the LQ model was that (borrowing the language of target theory for comparative purposes only) DNA (or a chromosome) was the target, and the hits corresponded to irreparable or misrepaired lesions produced by either one or two radiation tracks traversing the cell nucleus (Fig. 27-8). A comparison of the features and parameters of the target theory and LQ survival curve models is shown in Figure 27-9. To bridge the gap between the radiation responses of single cells grown in culture and tissues or tumors in a laboratory animal or human patient, several ingenious methods were developed to measure, or at least estimate, the radiation sensitivity of intact normal tissues and tumors in vivo.31–31 Some of these assays used the reproductive integrity of cells as an end point, similar in principle to the in vitro survival curve assay, but in which the animal essentially served as its own Petri dish. One classic example of an in vivo clonogenic assay is the spleen colony assay of Till and McCulloch,30 which originally was developed as a model system for the study of bone marrow transplantation. These authors determined that lethally irradiated mice could be “rescued” by a bone marrow transplant and that the transplanted, viable bone marrow stem cells were noted to form discrete nodules or colonies in the spleens of irradiated animals. By extension, then, a mouse’s spleen could be used as a pseudo Petri dish, with the number of clonogenic bone marrow colonies countable as a function of the radiation dose that the donated bone marrow received prior to transplantation. Assays such as this showed that the radiosensitivity of individual cells (tumor cells or normal cells) was largely unchanged whether the cells were irradiated in relative isolation in Petri dishes or as parts of a more complex tissue containing many different, interacting cell types in 3D contact. Two examples of nonclonogenic assays, one for normal tissues and one for tumors, are worth mentioning, especially because aspects of both are used routinely in the clinic, if not in the exact same way as the laboratory assay. One of the first nonclonogenic methods developed to assess normal tissue radioresponse was the skin reaction assay.32 (Pigs were used in the original studies because their skin is similar to that of humans in several key respects, although rodents have also been used.) An ordinate scoring system was used to quantify the severity of the skin reaction; for example, a skin score of “1” might correspond to mild erythema, whereas a score of “4” might correspond to confluent moist desquamation over more than half of the irradiated area. Then, when comparing different time, dose, and fractionation schedules, any combination that resulted in the same skin reaction score was assumed to correspond to an equivalent amount of cell killing. In this way, and by collecting information for different severities of skin reactions, a dose-response curve could be generated. A common nonclonogenic assay of tumor response to radiotherapy is the regrowth delay assay.33 In this assay, the tumor’s dimensions (or volume) are measured periodically as a function of time after irradiation, with the degree of tumor shrinkage assumed to be a reflection of the fraction of clonogenic tumor cells killed. Dose-response curves are generated by plotting the amount of growth delay (in days) as a function of radiation dose. As discussed previously, factors both intrinsic and extrinsic to the cell can alter its radiosensitivity. In addition to the intrinsic, genetic determinants of radiation sensitivity, other physical and chemical modifiers can also play important roles. The type of radiation used for treatment can be considered a physical modifier of radiosensitivity to the extent that high LET types of radiation (e.g., neutrons and heavy ions) are more biologically effective for a given unit of dose than low LET types (e.g., x-rays and electrons). Representative dose-response curves for radiations with different LETs are shown in Figure 27-10. In light of these differences in biological potency, the term relative biological effectiveness (RBE) has been coined to compare and contrast two radiation beams of different LET. RBE is defined as the ratio of doses of a known type of low LET radiation (historically, 250 kVp x-rays were the standard, but others can also be used) to that of a higher LET radiation to yield the same biological end point. RBE is highly variable and depends on several irradiation parameters, including the type of radiation, total dose, dose rate, dose fractionation pattern, and the biological effect being assessed. An example of how RBE values are obtained from cell survival curves is shown in Figure 27-11. Chemical modifiers of radiation response are also important, and perhaps the “chemical” of greatest significance in this regard is molecular oxygen, a potent radiosensitizer. Mechanistically speaking, the reason that well-oxygenated cells are more sensitive to radiation than cells relatively lacking in oxygen is that oxygen readily participates in the free radical reactions that occur in the micro- to milliseconds after irradiation and has the net effect of enhancing the radiation damage to cellular macromolecules. Poorly oxygenated cells experience less enhancement of radiation injury and are therefore more radiation resistant. This phenomenon is termed the oxygen effect. The relative resistance of poorly oxygenated cells compared with well-oxygenated ones can be expressed in terms of an oxygen enhancement ratio (OER). The OER is the ratio of doses to produce the same biological effect under low versus normal conditions of oxygenation. The OER typically ranges between 2.5 and 3.0 for large single doses of x-rays or γ-rays and 1.5 to 2.0 when multiple small dose fractions are used.34 Further, the oxygen effect is reduced or eliminated as the LET of the type of radiation increases; OERs of 1.5 to 2.0 are obtained for radiations of intermediate LET, and an OER of 1.0 (i.e., no oxygen effect) is obtained for high LET radiations. Representative radiation survival curves generated in the presence or relative absence of oxygen are shown in Figure 27-12. Hypoxic cell radiosensitizers, exogenous chemicals that mimic the radiation damage–enhancing effects of oxygen, have been used clinically in an attempt to combat the relative radiation resistance of tumors that contain a clonogenic fraction of poorly oxygenated, hypoxic cells. A second approach to the problem posed by tumor hypoxia is to combine radiotherapy with a drug that, rather than sensitizing hypoxic cells, kills them outright. These drugs are called “bioreductive” because their toxic effects only occur secondary to the reductive metabolism common in cells relatively lacking in oxygen.35,36 These drugs are discussed further later in this chapter. One of the first studies demonstrating an association between directly measured oxygen tension in tumors and clinical outcome was published in 1988 by Gatenby et al.37 An oxygen-sensing electrode was inserted into patients’ tumors (advanced squamous cell carcinomas of the head and neck), and multiple readings of partial pressure of O2 were taken at different depths along the probe’s track; the arithmetic mean pO2 value for each tumor positively correlated with local control rate, as did the tumor volume–weighted pO2 value. A high tumor oxygen tension was associated with a high complete response rate and vice versa. Several years later, comparable oxygen electrode results for uterine cervix cancers were obtained by Höckel et al.38 They made the important discovery that tumor hypoxia was a negative prognostic indicator in general, regardless of whether the patient had received radiation therapy, suggesting that radiation resistance was only one aspect of the hypoxia problem. A large meta-analysis of the relationship between tumor oxygenation status measured with oxygen electrodes and clinical outcome was published more recently for advanced head and neck tumors,39 with comparable findings of improved overall survival in patients whose tumors were less hypoxic. Another method of directly identifying and quantifying hypoxic cells in tumors is through the use of “hypoxia markers.” Both exogenous and endogenous markers for tumor hypoxia are available and can be studied in relation to each other, the tumor vasculature, or other “markable” features of the tumor microenvironment (e.g., the proliferation status of tumor cells). Exogenous markers consist of injectable drugs or chemicals that are bioreducible only under hypoxic conditions, causing them to bind to cellular proteins. These bound metabolites can be marked radioactively or with antibodies and can be visualized using several different techniques, such as autoradiography40 or fluorescence immunohistochemistry.41 Two exogenous markers studied extensively in both preclinical cell and animal systems, as well as in human patients, are pimonidazole hydrochloride42,43 and EF-5.44 Both are of the chemical class known as nitroimidazoles that were originally studied as possible radiosensitizers of hypoxic cells (e.g., see references 45 and 46, as well as the discussion later in this chapter) but were serendipitously found to also have the property of bioreduction and binding to cellular macromolecules under hypoxic conditions. This facilitated their use as hypoxia markers. Endogenous markers, on the other hand, consist of genes and proteins that are upregulated as part of the cellular stress/adaptive response to hypoxic conditions and whose expression levels can, with caveats, be used as surrogates for tissue oxygenation status.47,48 Commonly studied endogenous hypoxia markers include the hypoxia-inducible factor 1–alpha (HIF-1α, a transcription factor),48 the enzyme carbonic anhydrase IX (CA-9 or CAIX),49 glucose transporter–1 (GLUT1),52–52 and osteopontin.53 Figure 27-13 illustrates the potential of deriving “geographic” information about the extent and location of hypoxia in six different human tumors, using the exogenous marker pimonidazole hydrochloride. Simultaneously marking other tumor features, the location of blood vessels and proliferating cells in these examples, provides additional information about the tumor microenvironment. The concept of therapeutic ratio reflects the ability to estimate the likelihood of complication and tumor control in a given situation. At a given dose, these probabilities can be estimated (although usually not very accurately in clinical practice). Advances in radiation oncology can be made by moving the normal tissue complication curve to the right (as illustrated in Figure 27-14) so as to produce fewer complications at the same radiation dose, or by moving the tumor control curve to the left, so that less physical dose is needed to control the tumor (while also producing fewer complications).
Basics of Radiation Therapy
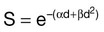 . The α/β ratio is a convenient metric for describing cellular radiosensitivity and has been adapted to describe the response of irradiated tissues as a function of time, dose, and fractionation.
. The α/β ratio is a convenient metric for describing cellular radiosensitivity and has been adapted to describe the response of irradiated tissues as a function of time, dose, and fractionation.
Overview of Radiation Physics
Interactions of Radiation and Matter
Photoelectric Effect
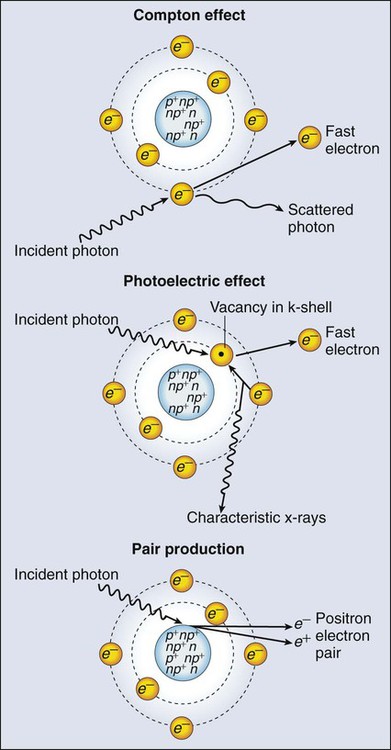
Compton Scattering
Pair Production
The Generation of Therapeutic Radiation
Linear Accelerators

Radioactive Sources
Isotope
Half-Life Average
Energy (keV)
PHOTON
226Ra
1620 y
830
137Cs
30 y
662
198Au
2.7 d
412
192Ir
73.8 d
370
125I
60 d
28
103Pd
16.97 d
21
Isotope
Half-Life
Maximum Energy (keV)
BETA
32P
14.3 d
1710
90Sr/90Y
28.5 y/2.7 d
550/2280
188W/188Re
69.4 d/17 h
350/2120
186Re
3.8 d
1070
62Zn/62Cu
9.3 h/9.7 min
660/2930
133Xe
5.2 d
360
131I
8.0 d
600
89Sr
50.5 d
1495
166Ho
26.8 h
1850
Delivery of Therapeutic Radiation
The Radiobiology of Radiation Therapy
Mechanisms of Radiation Damage to Cells
The Molecular Biology of Cellular Radiation Responses
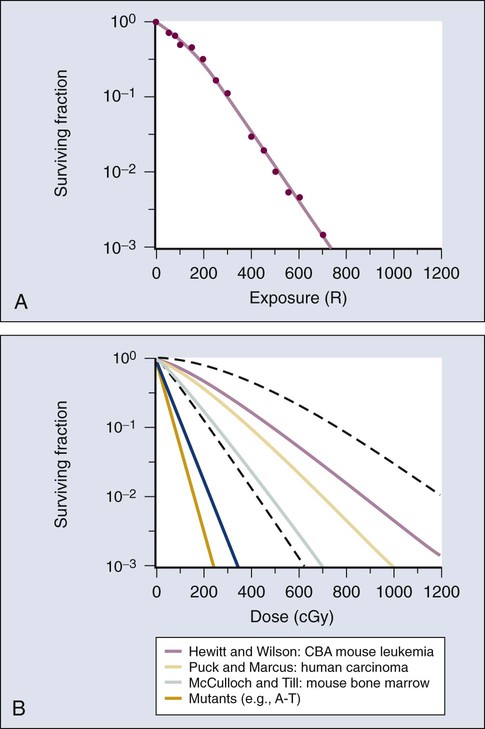
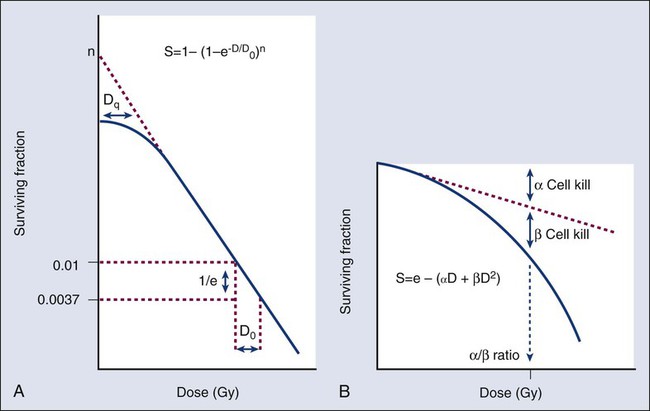
Cell Survival and Tissue Dose-Response Curves
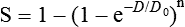
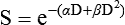
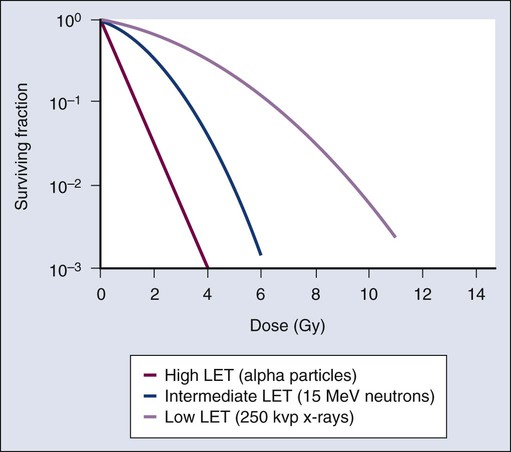
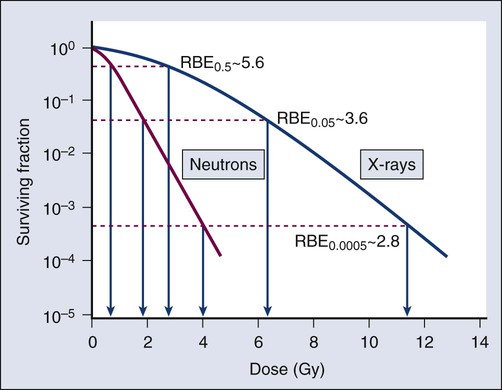
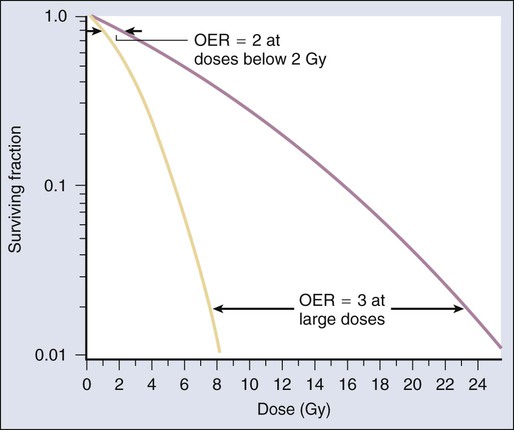
Modifiers of Radiation Sensitivity
Clinical Radiation Oncology
Therapeutic Ratio
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basics of Radiation Therapy