Key Points
- 1.
In malignant tumors, unregulated growth appears to result from a combination of loss of normal cell cycle controls and a failure of normal apoptotic mechanisms. Despite uncontrolled growth, malignant cell division does not appear to be more rapid than normal cell division.
- 2.
In general, as tumors grow, they display gompertzian growth characteristics: As the tumor mass increases in size, the time necessary to double its size also becomes progressively longer. Metastases generally have a shorter doubling time than the primary lesion.
- 3.
In late stages of tumor growth, only a few doublings in tumor mass may make a dramatic impact on the size of the tumor and the status of the patient.
- 4.
Chemotherapeutic agents work by first-order kinetics—that is, they kill a constant fraction of cells rather than a constant number.
- 5.
The effectiveness of any cancer treatment is limited by drug resistance, which may be intrinsic or acquired and may develop to one drug or to multiple drugs.
- 6.
Alkylating agents, such as the platinum compounds and cyclophosphamide, prevent cell division primarily by cross-linking strands of DNA.
- 7.
Antimetabolites, such as 5-flurouracil and methotrexate, act by inhibiting essential metabolic processes that are required for synthesis of purines, pyrimidines, and nucleic acids. These agents are typically S phase specific.
- 8.
Antitumor antibiotics, such as bleomycin and doxorubicin, generally work via DNA intercalation.
- 9.
Several chemotherapeutic agents, such as the taxanes, work via inhibition of normal microtubule function.
- 10.
Antiangiogenesis therapies, such as bevacizumab, are among the most common targeted agents.
- 11.
Hematologic toxicity and nausea and vomiting are the most frequently observed side effects in patients undergoing chemotherapy.
- 12.
Common antiemetic regimens include use of a 5-HT3 receptor antagonist with dexamethasone or aprepitant, a substance P/NK1 antagonist.
- 13.
The taxanes and platinum compounds are the most likely to cause hypersensitivity.
- 14.
Unlike reactions to taxanes, which typically occur within minutes of starting the initial dose, reactions to platinum agents typically do not manifest until several cycles have already been administered.
Historical Overview
The era of modern chemotherapy began in the 1940s with the Nobel Prize–winning work of Huggins and Hodges on the antitumor effect of estrogens in prostate cancer. This observation was followed in the mid-1940s by the investigation of nitrogen mustard, a byproduct of nitrogen gas used in World War I, for its effects against lymphomas and solid tumors. Between 1945 and 1965, a wide variety of chemotherapeutic agents was identified and studied, including actinomycin D, cyclophosphamide, the vinca alkaloids, 5-fluorouracil (5-FU), and the progesterones. In the 1970s, cisplatin was noted to exert significant antitumor effects against ovarian and testicular cancers, and tamoxifen was found to have activity against breast cancer for both adjuvant therapy and treatment of advanced disease. In the same decade, bleomycin, etoposide, and doxorubicin came into clinical use, and derivative compounds such as carboplatin, vinorelbine, and idarubicin were developed for their ability to achieve similar antitumor effects but with less hematologic toxicity. The 1980s and 1990s led to the widespread use of a host of new drugs, such as the taxanes (paclitaxel and docetaxel), ifosfamide, the topoisomerase inhibitors (topotecan and irinotecan), and nucleoside analogs (gemcitabine and capecitabine).
The growing number of agents in the chemotherapeutic armamentarium has been accompanied by advances in alternative dosing regimens; differing formulations using liposomal or polymer-based encapsulation; and varying schedules, sequences, and routes of administration. Fortunately, supportive therapies for gastrointestinal (GI) and hematologic toxicities have also evolved to include routine usage of 5-HT3 receptor antagonists (eg, ondansetron and granisetron) for antiemetic prophylaxis and colony-stimulating factors (eg, sargramostim and filgrastim) to allow for greater chemotherapeutic dose intensity.
General Principles
Chemotherapeutic agents are a crucial part of physicians’ armamentarium in the ever-broadening fight against cancer. Physicians can, with use of these drugs, ameliorate and sometimes even cure diseases that were usually fatal in the past. Until recently, in most cases, chemotherapy has been reserved for relatively late stages of the disease, but its increasingly successful use, particularly in the treatment of hematologic malignancies, suggests that chemotherapy should be administered earlier. All physicians and surgeons must understand the nature and use of cancer chemotherapy so they can make rational decisions about when it may be indicated. The outcome of cancer chemotherapy is not fully predictable, but the chances of remission can be improved by judicious selection of patients, careful assessment of the tumor’s growth pattern, and treatment of the neoplasm with the drug or drugs most likely to be effective. The clinical response to chemotherapy may be assessed using standard Response Evaluation Criteria in Solid Tumors defined by the National Cancer Institute ( Table 17.1 ).
| Evaluation | |
|---|---|
| TARGET LESIONS | |
| Complete response | Disappearance of all target lesions |
| Partial response | At least a 30% decrease in the sum of the longest diameter of target lesions, taking as reference the baseline sum longest diameter |
| Progressive disease | At least a 20% increase in the sum of the longest diameter of target lesions, taking as reference the smallest sum longest diameter recorded since the treatment started or the appearance of one or more new lesions |
| Stable disease | Neither sufficient shrinkage to qualify for partial response nor sufficient increase to qualify for progressive disease, taking as reference the smallest sum longest diameter since the treatment started |
| NONTARGET LESIONS | |
| Complete response | Disappearance of all nontarget lesions and normalization of tumor marker level |
| Incomplete response or stable disease | Persistence of one or more nontarget lesion(s) or maintenance of tumor marker level above the normal limits |
| Progressive disease | Appearance of one or more new lesions or unequivocal progression of existing nontarget lesions * |
* Although a clear progression of “nontarget” lesions only is exceptional, in such circumstances, the opinion of the treating physician should prevail, and the progression status should be confirmed later on by the review panel (or study chair).
Unfortunately, not all patients with cancer are amenable to chemotherapy. The suitability of a patient for treatment depends on at least three critical criteria:
- 1.
The nature of the neoplasm
- 2.
Its extent of spread or stage
- 3.
The patient’s clinical condition
Not all cancers are equally sensitive to drugs. Factors that determine a given tumor’s susceptibility include how the drug is distributed to the tumor, drug transport into the cell, whether a drug-sensitive biochemical pathway is present in the tumor cell, and the relative rates of intracellular activation and inactivation of the drug. A thorough knowledge of the cell cycle and growth kinetics is fundamental to understanding of the appropriate uses of chemotherapy.
Cell Cycle Control and Growth Kinetics
All living things have an inherent capacity to multiply, and they cease multiplication for various reasons. Control appears to be mediated by an unknown feedback mechanism, probably resulting from contact phenomena when cells are crowded together. Knowledge of growth patterns has aided in the derivation of chemotherapeutic principles. Strategies for therapy have evolved to take advantage of these differences in growth characteristics between normal and malignant tissues.
Normal tissues fall into three predominant categories: static, expanding, and renewing. Static populations of cells are generally well differentiated and after a period of proliferation in fetal life rarely undergo division during adult life. Examples of static tissues include neurons and skeletal muscle. Because of the rare incidence of cell division, these cells are unlikely to be injured by chemotherapies that target rapidly dividing cells. Expanding tissue populations are also usually inactive in adult life, but unlike static populations, they retain the ability to proliferate rapidly in response to stress or injury. Typical examples of expanding cells include hepatocytes and vascular endothelium. Last, the renewing cell populations are those that are constantly undergoing division, such as bone marrow and GI epithelium. Renewing tissues are most sensitive to injury by chemotherapeutic agents ( Table 17.2 ).
| Renewing (Rapid Proliferation) | Expanding (Slow Proliferation) | Static (Rare Proliferation) |
|---|---|---|
| Bone marrow | Lung | Muscle |
| Gastrointestinal mucosa | Liver | Bone |
| Ovary | Kidney | Cartilage |
| Testis | Endocrine glands | Nerve |
| Hair follicles | Vascular endothelium |
In malignant growth, cells do not cease multiplying when they reach a critical mass. This unregulated growth appears to result from a combination of loss of normal cell cycle controls and a failure of normal apoptotic mechanisms. Despite uncontrolled growth, malignant cell division does not appear to be more rapid than normal cell division.
In general, as tumors grow, they display gompertzian growth characteristics ( Fig. 17.1 ): As the tumor mass increases in size, the time necessary to double its size also becomes progressively longer. Thus, in the early phases of growth, tumor cells appear to grow exponentially, but as tumor mass increases, there is a progressive increase in the doubling time, although doubling times in humans may vary greatly. For example, whereas embryonal tumors and some lymphomas have relatively short doubling times (20–40 days), adenocarcinomas and squamous cell carcinomas have relatively long doubling times (50–150 days). Three explanations have been given for this prolonged volume-doubling time:
- 1.
An increase in cell cycle time (the time from one mitosis to the next)
- 2.
A decrease in the growth fraction (cells participating in cell division in the tumor)
- 3.
An increase in cell loss from tumor cells with insufficient nutrients and vascular supply

The gompertzian model has several important implications for cancer progression. First, metastases generally have a shorter doubling time than the primary lesion. If it is assumed that an exponential growth occurs early in the malignancy’s history and that the malignancy starts from a single cell, then a 1-mm mass will have undergone approximately 20 tumor doublings. A 5-mm mass (a size that is first recognizable on a radiograph) may have undergone 27 doublings. It follows that a 1-cm mass will have undergone 30 doublings, and a clinician will be pleased to have detected such an “early” lesion. Unfortunately, this “early” lesion has already undergone 30 doublings, with significant DNA change being possible. Using this rationale, clinical techniques that are currently available tend to recognize malignancies late in their growth, and metastatic disease may well have occurred long before there was obvious clinical manifestation of the primary lesion. Another implication from this kinetic information is that in late stages of tumor growth, a few doublings in tumor mass make a dramatic impact on the size of the tumor and the status of the patient. When a tumor becomes palpable (1 cm in diameter—30 doublings), only three more doublings will produce a very large tumor mass (8 cm in diameter).
The gompertzian model also has clinical implications that have guided a good deal of clinical chemotherapy research. As a mass responds to treatment (ie, gets smaller), the doubling time has been assumed to decrease as a consequence of a greater number of cells moving into cycle. This larger percentage of metabolically active cells would therefore increase the sensitivity of the neoplastic population of cell cycle–specific agents. This has led to the sequential use of cell cycle–nonspecific agents (eg, cyclophosphamide) to bring down the mass to be followed by cell cycle–specific agents (eg, methotrexate). Although these sequential combinations have been theoretically attractive, none has shown clear superiority in clinical trials. Another implication of the gompertzian growth concept is that metastases can be expected to be more sensitive to chemotherapy in general, and to cell cycle–specific agents in particular, than the primary tumor from which they arise. The smaller the size of the metastatic focus, the greater is the differential sensitivity. Therefore, the insensitivity of a primary tumor to a given drug regimen might not necessarily predict the response of metastasis to the same regimen.
The rationale for the use of drugs in the treatment of cancer is to achieve the selective killing of tumor cells. Underlying this rationale are the basic principles of the “cell kill” hypothesis first described by Skipper and associates. The following four principles were worked out in the L1210 leukemia model.
- 1.
The survival of an animal with cancer is inversely related to the number of cancer cells.
- 2.
A single cell is capable of multiplying and eventually killing the host.
- 3.
For most drugs, a clear relationship exists between the dose of the drug and its ability to eradicate tumor cells.
- 4.
A given dose of a drug kills a constant fraction of cells, not a constant number, regardless of the cell numbers present.
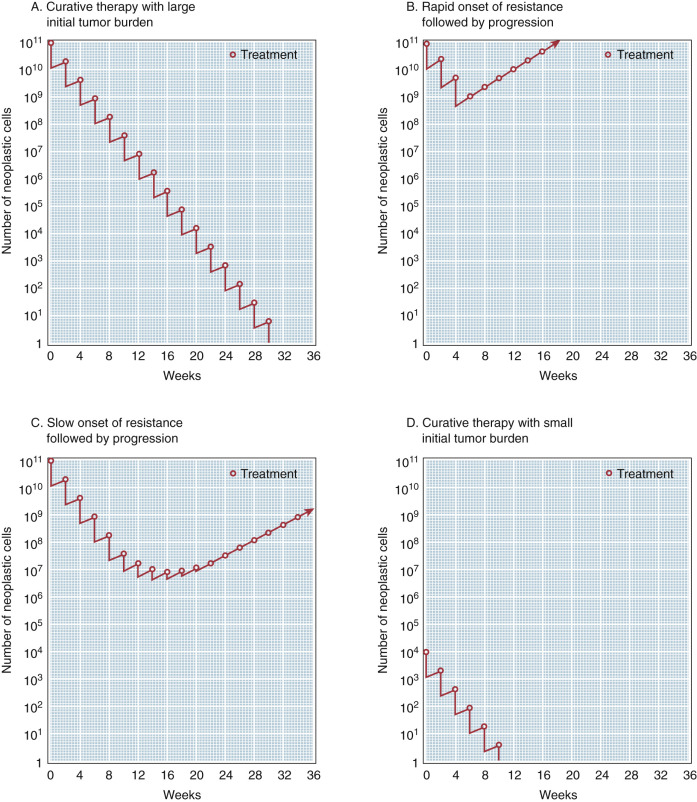
This fourth and most important principle implies that chemotherapeutic agents work by first-order kinetics—that is, they kill a constant fraction of cells rather than a constant number. This concept has important implications in cancer treatment. A single exposure of tumor cells to an antineoplastic drug may be capable of producing two to four logs of cell kill. With a common tumor burden of 1012 cells (1 kg), a single dose of chemotherapy will destroy a large number of cells but not be curative. Thus, there is a need for intermittent courses of chemotherapy to achieve the magnitude of cell kill necessary to eradicate the lesion. Clinically, first-order cell kinetics dictate that to eradicate a tumor population effectively it is necessary to either:
- •
Increase the total dose of the drug or drugs to the maximal limits tolerated by the host, or
- •
Start treatment when the number of cells is small enough to allow the destruction of the tumor at total doses of the drug that are reasonably tolerated.
The logical conclusion derived from this hypothesis is that the maximal opportunity for achieving cure exists during the early stage of disease. In the past, chemotherapy was generally reserved for the treatment of disseminated cancer; surgery and radiotherapy were treatments of choice for localized disease. However, this concept of “log kill hypothesis” provides a rationale for the philosophy of adjuvant chemotherapy, which assumes the presence of undetectable cell masses of 101 to 104 cells after the initial surgical therapy that are capable of producing tumor relapse. This small tumor burden is particularly vulnerable to effective chemotherapy.
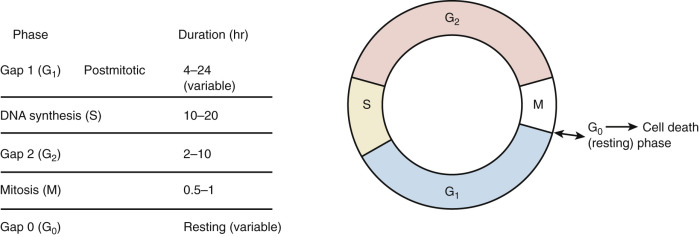
To better understand cell kinetics, it is imperative to visualize cell cycling. All dividing cells follow a predictable pattern for replication. The time that it takes a cell to complete one cycle of growth and division is termed its generation time . There are five basic phases ( Fig. 17.2 ). The G1 phase (G stands for gap and uncertainty as to purpose) lasts for a variable amount of time—usually between 4 and 24 hours. If this phase is prolonged, the cell is usually referred to as being in the G0, or resting, phase. The S phase is the phase of DNA synthesis and usually lasts between 10 and 20 hours. The G2 phase is a premitotic phase that lasts from 2 to 10 hours, and the M phase, when actual mitosis takes place, lasts between 0.5 and 1 hours. Tumors do not have faster generation times but have more cells in the active phases of replication than normal tissues. Normal tissues have a large number of cells in the G0 phase, wherein the cell is not actively committed to division or is “out of cycle.”
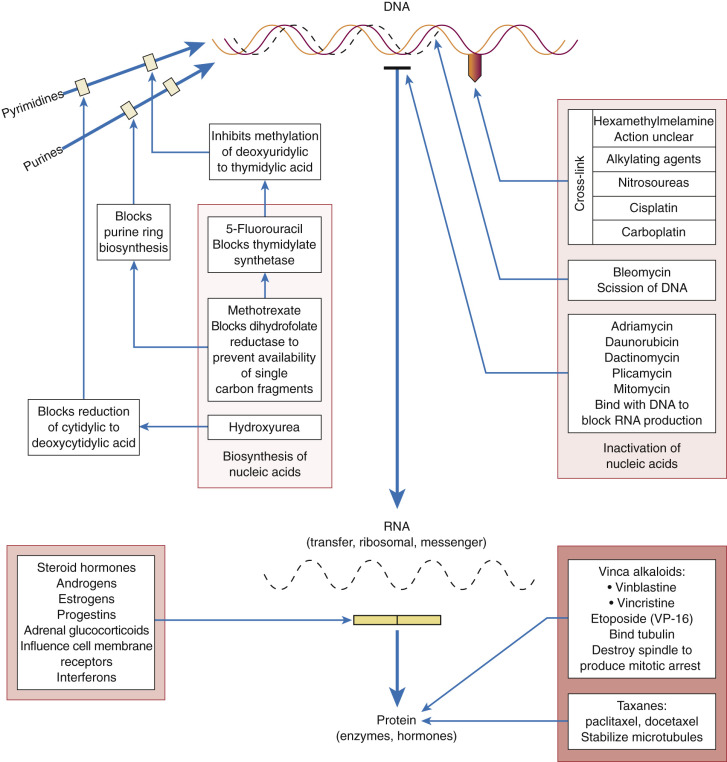
Some chemotherapeutic agents appear to act at several phases of the cell cycle ( Fig. 17.3 ). Alkylating agents appear to act in all phases from G0 to mitosis. They are called cycle-nonspecific agents. Drugs such as hydroxyurea, doxorubicin (Adriamycin), and methotrexate appear to act primarily in the S phase. Bleomycin appears to act in the G2 phase, and vincristine appears to act in the M phase. These drugs are called cycle-specific agents ( Table 17.3 ) because they act chemotherapeutically only on cells that are in a specific phase of a cell generation cycle. Steroids, 5-FU, and cisplatin have rather uniform activity around the cell generation cycle. In theory, if certain cancer therapeutic agents attack only cells that are dividing and more tumor cells are dividing than normal tissue cells, then by properly spacing the chemotherapeutic agent and combining agents that act in different phases of the cell cycle, one should be able to kill tumor cells in much greater numbers than normal cells. Kinetic studies in humans and animals suggest that tumors that have been cured by chemotherapy are those with large fractions of cells in the proliferative phase (eg, gestational choriocarcinoma and Burkitt lymphoma). The extent of the disease rather than the total mass of tumor is the most important factor when considering curative radiation or surgery, but in using chemotherapy, the total mass is most important. When tumor volume is reduced, the remaining tumor cells can begin to divide actively (they are propelled from the G0 phase into the more vulnerable cell generation cycle), thus rendering them susceptible to chemotherapy. These chemotherapeutic agents, as in radiation therapy, kill by first-order kinetics—that is, there is a reduction of the tumor population by a characteristic percentage, regardless of the actual number of tumor cells initially present ( Fig. 17.4 ). If the tumor burden is small, fewer cycles of chemotherapy may be necessary. One milligram of tumor usually consists of 106 cells. One cubic centimeter of tumor usually consists of 109 cells. Patient death usually occurs at 1012 cells.
| Phase Dependence | Type | Drugs |
|---|---|---|
| S phase dependent | Antimetabolite | Cytarabine |
| Doxorubicin | ||
| 5-Fluorouracil | ||
| 6-Mercaptopurine | ||
| Methotrexate | ||
| Hydroxyurea | ||
| Prednisone | ||
| M phase dependent | Vinca alkaloids | Vincristine |
| Vinblastine | ||
| Taxanes | Paclitaxel | |
| Docetaxel | ||
| Podophyllotoxins | Etoposide | |
| Teniposide | ||
| Epothilones | Ixabepilone | |
| G2 phase dependent | — | Bleomycin |
| G1 phase dependent | — | Corticosteroids |
Dynamics of Chemotherapy
The doubling time of a tumor depends on both generation time and cell death rate ( Fig. 17.5 ). One cannot assume a long generation time simply because a tumor enlarges slowly. Slow tumor growth can result from rapid generation time combined with a high cell death rate. For similar reasons, a small tumor discovered on radiographic or physical examination is not necessarily an early tumor; only serial studies to judge its growth rate will help establish its age. Bulky tumors (diameters >2–3 cm) enlarge more slowly than small ones because their cells, especially those of the inner core (farthest from the blood supply), have a long generation time. Competition for nutrients and other less-defined competitive pressures reduce the activity of the entire mass.
Successful chemotherapy of cancer requires a physiologic edge that can be exploited to differentially kill cancer cells but spare normal cells as much as possible. The more rapid growth rate of tumors and the increased synchronicity of tumor cells compared with normal tissues may be taken advantage of when designing therapeutic regimens. At any given time, comparatively large numbers of cancer cells will be in the DNA synthesis phase (S phase) of the cell cycle, the only time during which cycle-dependent agents (those inhibiting DNA synthesis) can act. Thus, short-term high-dose chemotherapy with agents affecting DNA synthesis, such as methotrexate, is most effective in killing rapidly dividing tumor cells with relative sparing of normal bone marrow elements. Unfortunately, bone marrow cells, the epithelial cells that line the GI tract, and hair follicles all have generation times comparable with those of tumors, and they are therefore vulnerable to compounds that inhibit DNA synthesis (see Table 17.2 ). However, compared with the more synchronously growing tumor cell population, only a few of the bone marrow cells are in their S phase at any given time, and this accounts for the selective toxicity of phase-dependent compounds. A course of therapy extending over a period of several days or even weeks may be required to kill a slow-growing tumor in which only a few cells are in the stage of DNA synthesis at any one time. Agents that do not depend on DNA synthesis for their effects (ie, cycle-nonspecific agents), such as alkylating agents, are most effective against bulky, slow-growing tumors. The cells remaining after treatment tend to divide more rapidly and are more susceptible to attack by cycle-specific agents. Thus, there is some flexibility in the interplay of chemotherapeutic agents.
The phenomenon of increased susceptibility of tumor cells during recovery from alkylating agents is the rationale for sequentially combining cycle-nonspecific and cycle-specific agents in many new regimens. If, in addition, drugs with different mechanisms of toxicity are combined, each drug can be given safely in the dose used when it is given alone. Each drug chosen for combination therapy should have antitumor activity when used alone. Whenever possible, intermittent courses of chemotherapy are used to allow restoration of normal cells if they were reduced in number by treatment. In cases in which an antidote to the chemotherapeutic agent is known—for example, leucovorin (citrovorum factor, folinic acid) for methotrexate—the antidote can also be given to hasten normal cell recovery. Of course, the danger of revitalizing sublethally injured tumor cells also exists and must be evaluated with each new treatment regimen. Although careful studies are needed to compare each new combination with the single agents concerned, the trend in chemotherapy is unquestionably toward exploitation of drug combinations used simultaneously and sequentially.
Pharmacologic Principles
Several general pharmacologic factors significantly affect the appropriate use of chemotherapeutic agents, including drug absorption, distribution, transport, metabolism, and excretion. These principles not only impinge on drug effectiveness but also dictate drug dose and schedule and how drugs are selected for use in combination. The effectiveness of a given regimen depends on optimizing the concentration x time (also known as the area under the curve [AUC]) at the site of tumor. Drug absorption influences route of administration, which in turn affects the AUC. Whether a drug is given orally, intravenously, intraarterially, intramuscularly, or intraperitoneally is also determined by patient acceptance, feasibility, and toxicity. Drug distribution and delivery to the tumor site also affects AUC. Factors such as drug binding (to albumin or to plastic catheters), lipid solubility, and membrane transport are critical in determining effectiveness of a given agent. Certain sites in the body, such as the brain and testes, represent pharmacologic sanctuaries where drug delivery is limited. Similarly, poor tumor perfusion because of necrosis or hypoxia may also impair drug delivery and concentration. Understanding of membrane transport mechanisms is also key: Certain drugs, such as 5-FU and mitomycin C, may enter cells through passive diffusion, but others, such as cisplatin and melphalan, require active transport.
Drug metabolism is often necessary to convert an inactive prodrug into the active form. One example is cyclophosphamide, which requires cytochrome P-450 activation before antitumor effects are possible. Agents requiring hepatic metabolism to active forms are not amenable to intraperitoneal or intraarterial administration. Conversely, metabolism and excretion of the active drug also affects AUC. The liver and the kidneys are responsible for the majority of drug elimination, although excretion in bile, stool, and respiration may also contribute in some cases. Organ dysfunction may result in increased drug toxicity and may require dose modification.
Drug Interactions
Although patients undergoing chemotherapy treatment may also receive a variety of other drugs for treatment of acute side effects or chronic medical conditions, few drug interactions are clinically significant. However, a few interactions are noteworthy. In particular, whereas doxorubicin and taxane agents are known to exert increased toxicity in the setting of impaired biliary excretion, the platinum compounds and methotrexate may cause increased toxicity in the setting of decreased renal function. Aspirin and sulfonamides are known to displace methotrexate from plasma proteins, and direct chemical interactions are noted between cisplatin and mannitol and mitoxantrone and heparin.
Drug Resistance
The effectiveness of any cancer treatment is limited by drug resistance, which may be intrinsic or acquired and may develop to one drug or to multiple drugs (pleiotropic resistance). It has been suggested that spontaneous mutation is a basis for drug resistance. This spontaneous mutation occurs rapidly in malignant tumors. This concept, the Goldie–Coldman hypothesis, has been applied to the growth of malignant tumors and has important clinical implications. The theory suggests that most malignant cells begin with intrinsic sensitivity to chemotherapeutic agents but develop spontaneous resistance at variable rates. Goldie and Coldman have developed a mathematic model that relates curability to the time of recurrence of the singly or doubly resistant cells. Assuming that there is a natural mutation rate, the model predicts a variation in the size of the resistant fraction in tumors of the same size and type, which depends on that mutation rate and the point at which the mutation develops. Thus, the proportion of resistant cells in any untreated tumor is likely to be small, and the initial response to treatment would not be influenced by the number of resistant cells. In clinical practice, this means that a complete remission could be obtained even if the resistant cell line were present. The failure to cure such a patient, however, would depend directly on the presence of these resistant cells. This model of spontaneous drug resistance implies that minimizing the emergence of drug-resistant clones requires that multiple effective drugs or therapies be applied as early as possible in the course of the patient’s malignant disease process.
In cell lines and animal models, resistance to specific drugs likely occurs via a wide variety of mechanisms, although only a few have been confirmed to be of clinical significance in human cancers. These include increase in proficiency of DNA repair, decrease in drug uptake or increase in efflux by cells, increased levels of or alterations in target enzymes, alterations in drug activation or degradation, gene amplification, and defective drug metabolism. These mechanisms are reviewed in Table 17.4 .
| Mechanism | Drugs |
|---|---|
| Increase in proficiency of repair of DNA | Alkylating agents, cisplatin |
| Decrease in cellular uptake or increase in efflux of drugs | Cisplatin, doxorubicin, etoposide, melphalan, 6-mercaptopurine, methotrexate, nitrogen mustard, vinblastine, vincristine |
| Increase in levels of “target” enzyme | Methotrexate |
| Alterations in target enzyme | 5-Fluorouracil, 6-mercaptopurine, methotrexate, 6-thioguanine |
| Decrease in drug activation | Cytosine arabinoside, doxorubicin, 5-fluorouracil, 6-mercaptopurine, 6-thioguanine |
| Increase in drug degradation | Bleomycin, cytosine arabinoside, 6-mercaptopurine |
| Alternative biochemical pathways | Cytosine arabinoside |
| Inactivation of active metabolites by binding to | Alkylating agents, cisplatin, doxorubicin sulfhydryl compounds |
| Decreased activity of topoisomerase | Camptothecins, doxorubicin, etoposide |
| Alteration of tubulin-binding sites | Vincristine, paclitaxel, ixabepilone |
| Increased damage tolerance | Alkylating agents, cisplatin |
Multidrug resistance also occurs via various mechanisms. Some experimental evidence in murine tumors suggests that one form of multiple-drug resistance relates to the ability of drug-resistant tumor cells to limit drug accumulation of structurally unrelated agents. This cross-resistance is seen most often with natural products (eg, doxorubicin, etoposide, paclitaxel, and vinca alkaloids); resistance to a single drug may confer a cross-resistance to structurally dissimilar drugs with different modes of action. This is the best studied mechanism for multidrug resistance and has been characterized involving the p-170 glycoprotein and its gene MDR1. Ling initially demonstrated the appearance of a P-glycoprotein with a molecular weight of 170 kD on the cell membrane. The appearance of pleiotropic drug resistance is associated with permeability of the cell to accumulate and retain antineoplastic drugs. It has been demonstrated that this P-glycoprotein is directly related to the expression of resistance, and cells that revert to the drug-sensitive state lose this membrane glycoprotein. DNA can be transferred from resistant cells to the sensitive cells, producing a transfer of pleiotropic resistance to unexposed cells.
Although best characterized, MDR1 is unlikely to be the most common mechanism for chemotherapy resistance among ovarian cancers, given that most do not express the MDR1 gene. Another mechanism for the multiple-drug resistance phenotype is seen among alkylating agents, cisplatin, and irradiation. Resistance in this group of agents has been linked to elevations in intracellular glutathione levels and is not associated with an overall measurable decrease in drug accumulation. Other transport proteins, including multidrug resistance–associated protein, have been identified that do not involve the p-170 glycoprotein pump. Furthermore, alterations in genes controlling apoptosis and growth arrest have also been cited. Although the relative importance of these separate mechanisms in ovarian cancer remains to be established, it seems most likely that in clinical situations various combinations of mechanisms are at work.
Calculation of Dosage
Dosages of chemotherapeutic agents are usually discussed in terms of mg/kg of body weight or mg/m 2 of total body surface area ( Table 17.5 ). Dosage based on surface area is preferable to that based on weight, because surface area changes much less during the course of therapy, allowing a more consistent absolute amount of drug to be given throughout therapy. Dosages per unit are also more comparable in adults and children ( Figs. 17.6 and 17.7 ), and the variation in total dose between very obese and very thin people is minimized. Dosage in experimental animals expressed as mg/m 2 is more easily related to that in humans. In adults, mg/kg can be converted with reasonable accuracy to mg/m 2 by multiplying by 40.
| Equation * | |
|---|---|
| Mostellar (m 2 ) | √Weight × Height/3600 |
| DuBois and DuBois (m 2 ) | (Weight 0.425 ) × (Height 0.725 ) × 71.84 |
| Haycock (m 2 ) | (Weight 0.5378 ) × (Height 0.3964 ) × 0.024265 |


Dose adjustments should be made for patients who are likely to have a compromised bone marrow reserve—that is, those older than 70 years of age, those who have received previous pelvic or abdominal irradiation, and those who have had previous chemotherapy. In these subsets of patients, the physician should consider beginning with a dose reduced by 35% to 50% and escalate up to a full dose with subsequent courses if initial doses are well tolerated. In a similar manner, any moderate to severe toxicity during the patient’s course of therapy should direct a reduction in future doses. The adverse effects criteria table used by the Gynecologic Oncology Group is included as Appendix B .
Dose adjustments are often required in patients receiving anticancer agents that are eliminated by the kidneys. These adjustments reduce the likelihood of overly high plasma drug concentrations and the attendant risk of serious renal toxicity. Several techniques have been used to assess renal function (glomerular filtration rate [GFR]) in individuals with cancer. The calculated creatinine clearance (Cr Cl) using serum creatinine is the most commonly used. The elimination of creatinine is primarily via glomerular filtration, although a small amount may be secreted in the renal tubules. Several studies have compared the different methods of estimating Cr Cl using a serum creatinine value. These methods are based on correlations of Cr Cl with age, body weight, serum creatinine, and creatinine metabolism. The most utilized methods follow.
Jelliffe Method
The Jelliffe method was used originally as a simple estimate of Cr Cl using serum creatinine, making minor adjustments in the calculation for female patients. The current Jelliffe formula takes into consideration age and renal function, and is as follows:
Cr Cl ( mL/min ) = ( 98 − ( 0.8 × ( age − 20 ) ) / ( serum creatinine in mg/dL ) ) × Patient ’ s BSA/1 . 73 m 2
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


