and Fabiana Gatti de Menezes2
(1)
Curitiba, Paraná, Brazil
(2)
São Paulo, São Paulo, Brazil
List of tables
Table 4.1 | Pharmacokinetic interactions | |
Table 4.2 | Pharmacodynamic interactions | |
Table 4.3 | Chemotherapy and enzyme complexes that are substrates with clinical significance | |
Table 4.4 | Enzymatic conditions to take into account in the choice of therapy and dose adjustment in neonate patients | |
Table 4.5 | Important interactions involving chemotherapy and P-glycoprotein | |
Table 4.6 | Relationship between ABC expression and resistance to anticancer therapy | |
Table 4.7 | Nonsteroidal anti-inflammatory drugs and enzyme complexes that they inhibit or induce | |
Table 4.8 | Steroidal anti-inflammatories and enzyme complexes/proteins for which they are substrates, or that they inhibit or induce | |
Table 4.9 | Drug interactions involving aprepitant and dexamethasone | |
Table 4.10 | Drug interactions involving aprepitant and chemotherapy | |
Table 4.11 | Drug interactions involving dexamethasone and chemotherapy | |
Table 4.12 | Drug interactions involving antacids and chemotherapy | |
Table 4.13 | Drug interactions involving antibiotic drugs and chemotherapy drugs | |
Table 4.14 | Drug interactions involving antifungals and antineoplastic drugs |
List of figures
Figure 4.1 | Toxicity linked to ABC family in antineoplastic therapy | |
Figure 4.2 | Mechanism of action of leucovorin (folinic acid) | |
Figure 4.3 | Mechanism of doxorubicin cardiotoxicity | |
Figure 4.4 | Mechanism of ifosfamide toxicity |
4.1 Basis of Drug Interactions
A drug interaction can change both the pharmacokinetics and the pharmacodynamics of a drug. Changes in pharmacokinetics may modify the absorption, degree of binding to plasma proteins, biotransformation, or excretion of the drug, and more than one alteration can occur concomitantly [6, 52, 53].
Of all hospital admissions in the United States, 2.8 % are for drug interactions and, according to a questionnaire of the American Society of Health-System Pharmacists, drug-drug interactions were classified as one of patient’s major concerns in a health service [33].
A multicenter cross-sectional retrospective study, conducted in the intensive care units of seven teaching hospitals in Brazil, analyzed the charts of 1,124 patients admitted in 2007; during 24 h of admission, 793 patients had at least one drug interaction, representing 70.6 % of patients. In total there were 2,299 drug interactions, of 353 types, in which 350 interactions were classified as drug – drug, and 3 as drug – parenteral nutrition, making an average of 2.92 interactions per patient. Of the interactions in the study, 0.1 % represented contraindicated interactions, 36.5 % serious interactions, 50.1 % moderate interactions, and 13.3 % represented low interactions [9].
Most drug interactions that occur in pediatric oncology are related to supportive care and are exacerbated due to individual variations, such as genetics and age [23].
4.1.1 Pharmacokinetic Interactions
Absorption: Drugs that accelerate gastric emptying tend to increase gastrointestinal absorption, and drugs that delay gastric emptying tend to decrease gastrointestinal absorption. Gastrointestinal motility, splanchnic blood flow, and the drug particle size and pharmaceutical form, as well as physical and chemical factors such as pH, may alter the absorption of drugs [23, 52, 104].
Distribution: An increase in free drug concentration may be caused by the displacement of the drug of its plasma binding sites or tissues (by a second drug); increased elimination follows, and this reduces the total drug concentration in plasma, resulting that free drug concentration is almost the same as that before the administration of the second drug. Before dynamic equilibrium occurs, however, there may be toxicity due to an increase in the concentration of the free drug. When the second drug that caused the displacement also decreases the elimination of the first drug, there may be high toxicity [23, 54].
The levels of serum albumin (and also the levels of other proteins), if decreased, release drugs to interact with tissues. Albumin production capacity depends on factors such as nutrition. Albumin is synthesized in the liver, and the exposure of hepatocytes to hepatotoxins interferes with this synthesis. Growth hormone, insulin, testosterone, and thyroid and adrenal cortical hormones influence the amount of albumin production [8, 54, 104].
Biotransformation: Antineoplastic drugs are mostly metabolized to by-products, which are less liposoluble, and excreted; the liver is the main organ metabolizer of both phase I reactions (oxidation, reduction, or hydrolysis) and phase II reactions (conjugation, methylation, sulfation, acetylation, glucuronidation, or glutathione conjugation). Oxidative metabolism (phase I) usually occurs through the cytochrome P450 enzymatic system, which consists of approximately 100 isoenzymes, such as CYP3A4 (the most abundant enzyme in the liver); CYP3A4 accounts for approximately half of the drug metabolic activities of all cytochrome P450 enzymes [23, 104].
Metabolism by enzymatic induction: Many drugs induce drug-metabolizing enzymes; over 200 drugs cause enzyme induction. Typically, the inducer drug is a substrate for the enzyme, and one of the results of induction is the early elimination of the drug in the body. This may result in no treatment occurring [6, 46].
Metabolism by enzyme inhibition: Enzyme inhibition, especially of the P450 system, decreases the metabolism of drugs metabolized by those enzymes, increasing their effect [5, 54, 104].
Excretion: A drug can change the binding of another drug to plasma proteins, subsequently changing the drug’s filtration. Elimination can be altered by the inhibition of tubular secretion, or by alteration on urinary pH, as well as an alteration on urinary flow; both factors may operate at the same time, as well as only one of these factors can be modified [6, 53, 54, 104].
Table 4.1
Pharmacokinetic Interactions
Absorption | Accelerates gastric emptying: Increases gastrointestinal absorption. |
Distribution | A drug can displace another drug of its binding site, increasing the concentration of free drug. Later, dynamic equilibrium occurs resulting from the increased elimination, but before that can occur, there may be toxicity due the excess of free drug. If the drug that caused the displacement also decreases the removal of the first drug, there can be high toxicity [23, 54, 104]. The levels of serum albumin and other proteins, if decreased, release the drugs so that they are free to interact with tissues [23, 54, 104]. |
Biotransformation by enzyme induction | |
Biotransformation by enzyme inhibition | |
Excretion |
4.1.2 Pharmacodynamic Interactions
Pharmacodynamic interactions are often expected if the mechanism of action of medicines used is taken into account. These interactions occur by several different mechanisms; an antagonist of a given receptor may decrease the effectiveness of the same receptor agonist. Pharmacodynamics can be changed by competition for the receptor, and can also be changed by another medicine with a mechanism of action in another receptor [6, 52].
Effects of pharmacodynamic interactions: Most drugs are studied in young or middle-aged adults and there is little information on pharmacodynamics and pharmacokinetics in children. Because the pharmacokinetics and pharmacodynamics may be different in children, adjustment of the dose and posological schedule is generally required to achieve the desired therapeutic effect without risk [6].
In childhood, because the disposition of drugs does not vary linearly according to weight or body surface area, there is no trusted formula for calculating a safe and effective dose for children using the adult dose. It must be taken into account that pharmacokinetic variability tends to be higher in periods of physiological change, such as in newborns, premature infants, and individuals at the stage of puberty [6].
Addition: Drugs used together have additive pharmacological activity, resulting in a sum equal to some of the pharmacological effects of each alone.
Synergism: This is similar to the additive effect, but the final effect is greater than the sum of the effects of the individual agents.
Potentialization: This is an increase in the toxicity of an agent when used concomitantly with another non-toxic agent, causing a greater effect of the first agent.
Antagonism: The agent acts as an antidote to another agent.
Physiological or Functional Antagonism: The drugs produce opposite effects on the same function.
Chemical Antagonism: Produces chemical reaction between compounds, inactivating any of them.
Table 4.2
Pharmacodynamic interactions
Additive effect | |
Synergistic effect | |
Potentialization effect | |
Functional or physiological antagonism | |
Chemical antagonism | |
Processing antagonism |
4.2 Drug: Drug Interactions
4.2.1 Drug Interactions (Pharmacokinetics) Involving CYP450
Several P450 enzymes are located in the inner mitochondrial membrane with catalytic sites exposed on the matrix. Another important family of cytochrome P450 enzymes is found in the endoplasmic reticulum of hepatocytes. Many of the substrates of P450 enzymes (in the endoplasmic reticulum) are xenobiotics (compounds synthesized industrially and not found in nature). Through hydroxylation by cytochrome P450 enzymes, hydrophobic compounds become more water soluble and can be excreted by the kidneys. Metabolism by P450 enzymes limits the lifetime of a drug in the bloodstream and thus limits its therapeutic effect [46].
Drug toxicity is also frequently a result of enzymatic conversion, in which the drug molecule is converted into a reactive metabolite [54].
The concomitant administration of CYP450 inducers and chemotherapeutic drugs that are catabolized by cytochrome P450 enzymes may increase the clearance of the chemotherapeutic drug, increasing the risk of relapse and reducing the drug’s efficiency; administering these chemotherapy drugs together with CYP450 enzyme inhibitors can decrease clearance and increase toxicity [23].
Competitive inhibition occurs when drugs are substrates of the same CYP isoform; this inhibition is dose-dependent and significant when the isoform is saturated. After discontinuation of the drug that interacts, competitive inhibition is rapidly converted. Non-competitive inhibition occurs when the drug interaction destroys the CYP isoform; after discontinuation of the drug that is interacting, non-competitive inhibition takes days or weeks to be converted, because of the need for new synthesis [23].
Table 4.3
Chemotherapy and enzyme complexes that are substrates with clinical significance
CYP 2A6 | CYP 2B6 | CYP 2C8 | CYP 2C9 | CYP 2C19 | CYP 2D6 | CYP 2E1 | CYP 3A4 | |
|---|---|---|---|---|---|---|---|---|
Brentuximab | Substrate | |||||||
Busulfan | Substrate | |||||||
Cyclophosphamide | Substrate | Substrate | ||||||
Dacarbazine | Substrate | |||||||
Docetaxel | Substrate | |||||||
Doxorubicin | Inhibitor | Substrate | Substrate | |||||
Etoposide | Substrate | |||||||
Ifosfamide | Substrate | Substrate | Substrate | |||||
Imatinib | Inhibitor | Substrate Inhibitor | ||||||
Irinotecan | Substrate | Substrate | ||||||
Lomustine | Substrate | |||||||
Paclitaxel | Substrate | Substrate | Substrate | |||||
Vinblastine | Substrate | |||||||
Vincristine | Substrate | |||||||
Vinorelbine | Substrate |
Taking into account that pharmacokinetics and pharmacodynamics would vary more in periods of physiological change, the professionals involved in cancer therapy should be aware of the divergence of enzyme concentrations in these phases. At birth, many drug-metabolizing enzymes are expressed at low concentrations; after birth occurs, specific isoenzymes are induced. CYP2E1 and CYP2D6 appear on the first day after birth; and CYP3A4 and the CYP2C subfamily arise after a week. CYP2A1 is expressed 1–3 months after birth. CYP3A7 is present only in fetal livers. To adjust doses by weight or body surface area, we should take into account that the hepatic metabolism of drugs after the neonatal period is generally faster than in adults. Renal elimination of drugs, in general, is also reduced during the neonatal period, and full-term newborns have pronounced reductions in glomerular filtration rate (2–4 mL/min/1.73 m2), while premature newborns tend to have further reduced renal function; therefore, the dose regimens of some drugs for newborns must be reduced to avoid the toxic accumulation of drugs. Glomerular filtration rate gradually increases to adult levels (corrected based on body surface area) from ages 8 to 12 months [6].
Table 4.4
Enzymatic conditions to take into account in the choice of therapy and dose adjustment in neonate patients
CYP2E1 | Appears on the first day after birth |
CYP2D6 | Appears on the first day after birth |
CYP3A4 | Appears 1 week after birth |
CYP2C | Appears 1 week after birth |
CYP2A1 | Appears 1–3 months after birth |
Hepatic metabolism | Accelerated after the neonatal period |
Renal elimination | Decreased in the neonatal period |
Glomerular filtration rate | Decreased in newborns; premature infants have a more decreased glomerular filtration rate. Increases to adult levels (based on adjusted body surface area) from 8 to 12 months |
4.2.2 Drug Interactions (Pharmacokinetics) Involving Binding Proteins and Affinity for Plasma Proteins
These drug interactions are important in oncology, due to the narrow therapeutic index of chemotherapeutic drugs [23].
Plasma proteins make up more than 70 % of plasma solids [47].
Drugs with high affinity for plasma proteins may be displaced by other drugs with a higher affinity for the protein; this may increase the concentration of the free form of the first drug, thereby increasing the toxic effects [23].
4.2.2.1 α1-Acid glycoprotein (AAG)
AAG is present in plasma at 100 times lower concentrations than albumin. There is evidence that AAG participates in normal coagulation, tissue repair, and immunobiological processes. It is a naturally acid protein, due to the high amount of sialic acid, and it also has a low pKa. Acid and basic drugs can bind to this protein, and it has a site (site I) that binds acid drugs such as warfarin. Basic drugs, such as phenylbutazone, bind in site II. AAG is responsible for the greatest amount of difference in plasma protein binding between individuals. The variability of AAG amounts is vast between healthy and sick patients. AAG contributes significantly to the binding of acid drugs, while the capacity of albumin, or its affinity for binding, is also high for acid drugs.
The binding of basic drugs to AAG is variable because of the variability in the amount of AAG in individuals, also because this protein has high affinity and low capacity on its binding site, and because this protein may be easily saturated by increases in the concentration of the linker drug. Some basic drugs that bind significantly to AAG and are widely used in children are metoclopramide and prednisolone [55, 104].
4.2.2.2 Albumin
Albumin is the most important plasma protein and its concentration in healthy individuals does not vary significantly. Albumin is responsible for the major part of the total binding of basic drugs in plasma; propranolol, for example, binds to albumin in an amount of 50 % of this drug present in plasma. Because this protein tends to have low affinity for basic drugs and high capacity for binding to this kind of drug, changes in albumin concentration resulting in marked changes in drug binding are not common; however, significant changes in the amount of plasma albumin may trigger a remarkable change in the binding of albumin to basic drugs. Hyperalbuminemia is rare, but hypoalbuminemia can occur in cases of severe liver disease or severe kidney disease, especially in nephrotic syndrome [55, 104].
4.2.3 Drug Interactions (Pharmacokinetics) Involving Transport Proteins
4.2.3.1 P-glycoprotein
P-glycoprotein is a carrier protein that is located on the cell membranes of various tissues. P-glycoprotein removes chemical compounds from the cell and carries them, in the circulation, for elimination; it does not metabolize drugs, but carries the drugs to places that expose them to metabolic mechanisms such as the CYP450 system [23].
Drug | P-glycoprotein | Drug interactions |
|---|---|---|
Brentuximab vedotin | Substrate | The effect of brentuximab vedotin can be increased by P-glycoprotein inhibitor drugs. The effect of brentuximab vedotin can be reduced by P-glycoprotein inducer drugs. |
Doxorubicin | Substrate/inducer | The effect of doxorubicin can be increased by P-glycoprotein inhibitor drugs. The effect of doxorubicin can be reduced by P-glycoprotein inducer drugs. Doxorubicin may decrease the effect of P-glycoprotein substrate drugs. |
Daunorubicin | Substrate | The effect of daunorubicin can be increased by P-glycoprotein inhibitor drugs. The effect of daunorubicin can be reduced by P-glycoprotein inducer drugs. |
Etoposide | Substrate | The effect of etoposide can be increased by P-glycoprotein inhibitor drugs. The effect of etoposide can be reduced by P-glycoprotein inducer drugs. |
Idarubicin | Substrate | The effect of idarubicin can be increased by P-glycoprotein inhibitor drugs. The effect of idarubicin can be reduced by P-glycoprotein inducer drugs. |
Irinotecan | Substrate | The effect of irinotecan can be increased by P-glycoprotein inhibitor drugs. The effect of irinotecan can be reduced by P-glycoprotein inducer drugs. |
Methotrexate | Substrate | The effect of methotrexate can be increased by P-glycoprotein inhibitor drugs. The effect of methotrexate can be reduced by P-glycoprotein inducer drugs. |
Paclitaxel | Substrate | The effect of paclitaxel can be increased by P-glycoprotein inhibitor drugs. The effect of paclitaxel can be reduced by P-glycoprotein inducer drugs. |
Vincristine | Substrate | The effect of vincristine can be increased by P-glycoprotein inhibitor drugs. The effect of vincristine can be reduced by P-glycoprotein inducer drugs. |
Vinblastine | Substrate/inducer | The effect of vinblastine can be increased by P-glycoprotein inhibitor drugs. The effect of vinblastine can be reduced by P-glycoprotein inducer drugs. Vinblastine may decrease the effect of P-glycoprotein substrate drugs. |
4.2.3.2 ATP – Binding Cassettes
ABC transporters are so called because all of them have a domain that binds to ATP (ATP – binding cassettes). They form a large family of multidrug transporters that have two transmembrane domains with six helices and they transport a variety of ions, amino acids, vitamins, steroid hormones, and bile salts through the plasma membrane [48].
The expression of ABC is related to resistance to antineoplastic therapy, changes of antineoplastic drugs, and the toxicity of chemotherapy. Heterogeneity has been described in ABC genes, and some of this heterogeneity are related to changes in the pharmacokinetics and pharmacodynamics of chemotherapeutic drugs. Due to the multiple variations in transporting function and substrates, mutations in the genes and codification of these carriers can contribute to genetic disorders such as cystic fibrosis, neurological disorders, and anemia.
Based on phylogenetic classification, ABC proteins can be divided into seven subfamilies, with designations of ABCA to ABCG [34].
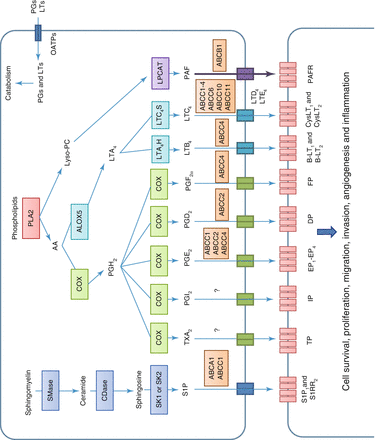
Table 4.6
Relationship between ABC expression and resistance to anticancer therapy
ABC subfamily | Genetic localization | Interaction in chemotherapy or cancer |
|---|---|---|
ABCB1 | 7p21 | Multiple drug resistance; tumor prognostic factor |
ABCC1 | 16p13.1 | Multiple drug resistance; tumor prognostic factor |
ABCC3 | 17q21.3 | Multiple drug resistance in lung cancer and other cancers |
ABCC4 | 13q32 | Transport of analogous nucleoside |
ABCC5 | 3q27 | Transport of analogous nucleoside, resistance |
ABCC6 | 16p13.1 | Resistance to anthracyclines and epipodophyllotoxins |
ABCG2 | 4q22 | Resistance to methotrexate, anthracyclines, topotecan, irinotecan, and mitoxantrone |
Fig. 4.1




Toxicity linked to ABC family in antineoplastic therapy. SMase Sphingomyelinase, CDase Ceramidase, SK1 and SK2 Sphingosine – kinase 1 and 2, S1P 1 and SRR 2 Sphingosine-1-phosphate receptors 1 and 2, PLA2 Phospholipase A2, AA Arachidonic Acid, Lyso-PC Lysophosphatidylcholine, COX Cyclo-oxygenase, ALOX5 5 – lipo – oxygenase, PGH 2 Prostaglandin H2, LT Leukotriene, LTA 4 H Leukotriene-A4-hydrolase, LTC 4 S Leukotriene-C4-synthase, LPCAT Lysophosphatidylcholine acetyltransferase, TX Thromboxane, PAF Platelet activating factor, TP Thromboxane receptor, IP Prostacyclin receptor, EP 1 -EP 4 Prostaglandin E receptors 1-4, DP Prostaglandin D2 receptor, FP Prostaglandin F receptor, BLT 1 and BLT 2 Leukotriene B4 receptors 1 and 2, CysLT 1 and CysLT 2 Cysteinyl leukotriene receptors 1 and 2, PAFR Receptor of platelet activating factor (Source: Based on information from Fletcher et al. [20])
Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
