Fig. 15.1
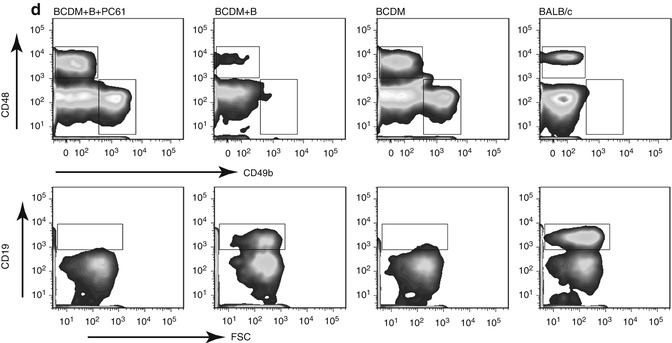
Tumor growth, CTL activity, IFN-γ expression; a comparison in B cell-depleted mice (BCDM), B cell-reconstituted BCDM, and immunocompetent wild-type (WT) mice. Mice were subcutaneously injected with 106 EMT-6 mammary tumor cells. For B cell-reconstituted BCDM, adoptive B cell transfers were done at day −7, 0, and +7. Spleens were harvested and processed 30 days post implantation. (a) Tumor growth following EMT-6 injection: five mice/group, mean tumor volume ± SEM. *p < 0.05; **p < 0.01. (b) CTL assay: EMT-6 tumor cells were treated with mitomycin C then cocultured for 7 days with splenocytes at an 8:1 splenocyte:tumor ratio. Using Lympholyte-M (Cedarlane), splenocyte effector cells were harvested then cocultured with mitomycin-C-treated Cr51-labeled EMT-6 at indicated E:T ratios for 4 h. Four mice/group, mean ± SD and 51 chromium release measured and percent lysis calculated. (c) Expression of IFN-γ in CD8+ T cells from representative mice in each group. CD8+ T cells were purified from splenocytes 30 days post tumor implantation then treated with PMA/ION for 4 h in the presence of GolgiStop (BD Pharmingen) and stained for CD8 followed by intracellular IFN-γ staining and analyzed by flow cytometry. (d) CD45+ tumor-infiltrating lymphocytes in tumor tissue from B cell-depleted mice (BCDM), B cell-reconstituted BCDM, and immunocompetent wild-type (WT) BALB/c mice with or without anti-CD25 antibody (PC61) treatment (PC61 administered on day −7 and day 0 relative to tumor implantation in treated group) were analyzed for CD4, CD8, CD49b, CD19, and Foxp3 expression. After 30 days post EMT-6 tumor implantation in each group with or without PC61 treatment, tumor was digested using Collagenase D/DNase. Dead cells, tumor cells, and red blood cells were removed using Histopaque-1077 (Sigma-Aldrich). Representative flow cytometry data for mice in each treatment group is shown using antibodies for CD8+ T cells, NK cells (CD49b), and/or CD19 + T cells as indicated [7] (Adapted from Zhang et al. [7] with kind permission from Springer Science and Business Media)
B cells can also modulate responses to vaccination in several murine tumor models. Using a secreted gp96-Ig heat shock protein-based vaccine in the LLC-OVA tumor model, Podack and coworkers demonstrated that rejection of established tumors required frequent and repeated vaccination in the presence of B cells, but in the absence of B cells, a single vaccine administration could elicit antitumor response resulting in rejection [10, 11]. Similarly, in a model for immunization with melanoma-associated antigens, using adenovirus encoding gp100 or murine TRP-2, single immunizations were incapable of inhibiting tumor growth in wild-type mice but could prevent the growth of B16 tumors in BCDM [12]. B cells may also serve to modulate and suppress antitumor effects of cytokines such IL-15. BCDM showed complete recovery in response to a combination treatment with cyclophosphamide and IL-15, whereas wild-type mice showed partial responses [13]. Collectively, these observations suggest a qualitative difference between BCDM and wild-type mice, which results in an impaired antitumor immune response in wild-type mice relative to BCDM, due to the suppressive effects of B cells.
In other studies, B cells were observed to play a critical role in the pathogenesis and development of tumors in response to chemical carcinogenesis. In a study using 7,12 dimethylbenz(a)anthracene (DMBA) and tetradecanoyl phorbol acetate (TPA) as a means of inducing papillomas in the skin of mice, it was found that both B cells and TNF-α were required for full-blown development and induction of papillomas. Transfer of normal B cells from DMBA-TPA-treated wild-type mice to TNF-α (−/−) mice would restore papilloma development to levels seen in the absence of B cells. B cell transfer from TNF-α (−/−) mice did not mediate this effect. In this model, resistance to papilloma development in TNF-α (−/−) mice was associated with an increase in interferon gamma (IFN-γ)-producing CD8+ T cells, as well as a reduction in IL-10-producing B-regulatory cells [14]. Hence, in that model B cells appear to be critical for tumor development and carcinogenesis, and the effects of B cells in modulating antitumor immunity appear to be complex. The relationship between B cell-mediated secretion of TNF-α and carcinogenesis is unclear; nonetheless, it is interesting to speculate that B cells fostered both an inflammatory environment and immunosuppressive aspect conducive to the development of papillomas. Collectively, it would appear that B cells play an important role in modulating antitumor responses in several murine systems.
The role of B cells in carcinogenesis may be complex. In a model of inflammation-associated epithelial carcinogenesis, in K14-HPV16 mice, the progression of malignancy appears to be dependent upon B lymphocytes [15]. Adoptive transfer of B lymphocytes and/or serum from HPV16 mice into B cell-deficient HPV16 mice restored progression from premalignancy to frank malignancy. The role of B cells in this case was somewhat unclear; however, more extensive innate immune cell infiltration was noted in premalignant tissue in the presence of B cells, which appears to enhance the establishment of a chronic inflammatory state, required for initiation of carcinogenesis. The fact that carcinogenesis was impeded by the absence of B cells suggests that in this model, B cells may either have been required for the establishment of chronic inflammatory state or, alternatively, the presence of B cells allowed the progression of carcinoma, perhaps due to its inhibitory effects on an adaptive immune response. Hence, the role of B cells may be complex and context dependent relating to specific mechanisms of carcinogenesis and immunogenicity of resultant tumors [16].
15.2 Mechanisms Underlying B Cell Modulation of Antitumor Immune Response
A variety of mechanistic explanations have been provided to explain observations of decreased antitumor immunity in the presence of B cells. One explanation that has been considered is that the presentation of tumor antigens by B cells may favor Th2-type T cell responses while diminishing Th1 response. Conversely, in the absence of B cell antigen presentation, T cell responses may tend toward Th1 response [17, 18]. Why B cell presentation of antigen may favor Th2-type responses is still a matter of speculation.
B cells may also differentiate along pathways in which they preferentially secrete Th1- or Th2-type cytokines. B cells which preferentially secrete IFN-α and IL-12 tend to augment Th1 response and have been labeled as Be1 cells, while B cells secreting IL-4 and IL-5 have been designated Be2 cells [19] and tend to support Th2 differentiation. Differentiation along Be1 and Be2 pathways has been well demonstrated following infection with pathogens that preferentially induce Th1- or Th2-type immune responses [20]. IL-4 and IL-4Rα receptor expression on B cells may result in preferential Be2 differentiation [21]. Be2 cells may skew differentiation along with Th2 pathways through elaboration of Th2-type cytokines. Whether tumor-infiltrating B cells are differentiated along Be1 or Be2 pathways in murine systems remains to be elucidated; nonetheless, it is conceivable that Be1 or Be2 differentiation could serve as a means of modulating antitumor immune responses by B cells, as has been observed in relation to several pathogens.
15.3 B Cells and the Role of Tregs
CD4+CD25+FoxP3+ Tregs have increasingly been identified as a means of suppressing CD8+ T cell responses. We and other laboratories have demonstrated that B cells may partially regulate the expansion of CD4+CD25+FoxP3+ T cells in both autoimmune and tumor settings [8, 22–28]. For example, in response to an antigenic challenge with ovalbumin coupled to a cholera toxin B subunit, the combination of B cell antigenic presentation and B cell elaboration of TGF-β and IL-10 appeared to induce Treg expansion and proliferation [23]. In another model using myelin oligodendrocyte glycoprotein (MOG) peptide as an antigen coupled with cholera toxin B, a similar expansion of Tregs was noted and served to suppress the development of experimental autoimmune encephalomyelitis [29].
In the EMT6 murine mammary tumor model in our laboratory, increased numbers of Tregs were noted following tumor inoculation in both BCDM and wild-type mice, and Tregs obtained from wild-type mice appear to have enhanced inhibitory function relative to Tregs isolated from BCDM suggesting conditioning of Treg function by B cells. Adoptive transfer of B cells into BCDM resulted in a marked increase of CD4+CD25+FoxP3+ Tregs compared to BCDM. The increase in Tregs correlated with enhanced tumor growth following B cell reconstitution [7, 8]. Interestingly, depletion of Tregs using an anti-CD25 antibody abrogated the growth of tumors despite the adoptive transfer of B cells into BCDM. Increased tumor growth was associated with diminished CD8+ T cell cytolytic response, as well as a decrease in CD8+ T cell and NK cell infiltration into tumors (Fig. 15.1). As previously mentioned, reconstitution with IL-10(−/−) B cells also facilitated EMT6 growth; hence, this Treg expansion and function did not appear to be contingent on B cell elaboration of IL-10 [7]. It would therefore appear that a variety of B cell regulatory subsets may affect response depending upon the tumor context and the overall nature of the immune response.
Other molecules that have been implicated in playing an important role in the development of Tregs are GITR and GITR ligand (GITR-L), co-stimulatory molecules belonging to the TNF superfamily. A variety of immune effector cells including B cells, NK cells, and CD8+ T cells, as well as Tregs express GITR following activation. GITR-L is expressed on a variety of cells including B cells and dendritic cells, endothelial cells, and others in a mouse [30]. In a mouse model of experimental autoimmune encephalomyelitis, B cells appear to regulate the number of CD4+CD25+FoxP3+ Tregs in the central nervous system through GITR-GITR-L interactions [31]. It is not known whether GITR-L expressing B cells also play an important role in the regulation of antitumor immunity.
A variety of investigators have demonstrated important regulatory role for B cells in autoimmune diseases including rheumatoid arthritis, systemic lupus erythematous (SLE), and inflammatory bowel disease [32–35]. In many of these systems, B-regulatory cell function appears to be mediated by IL-10. IL-10-producing B-regulatory cells (B10) were initially described in mice by Tedder and his group [36]. A variety of immunophenotypic markers serve to distinguish the IL-10+ subset. The so-called B10 cells appear to be important negative regulators of autoimmunity in mouse disease models. In a murine model for EAE, adoptive transfer of B10 cells prevents onset of disease [36]. B10-like cells have also been implicated in SLE in man [37]. The B10 subset appears to be able to skew T cell differentiation away from the Th1 pathway and has been implicated in the pathogenesis of SLE. However, in several murine tumor models, IL-10 appears to be less important for B cell-mediated immune suppression, and whether B10 cells have played a role in immune suppression in human tumors remains speculative.
A variety of other B cell subsets with regulatory function have been described including CD1d+ marginal B cells, transitional marginal and precursor B cells as well as CD5+ CD1d+ cells [32]. Gallipeau and coworkers also described the induction of regulatory B cell subset capable of attenuating autoimmune encephalitis in a murine model through administration of a GM-CSF-IL-15-fused cytokine or fusokine [38].
In addition to IL-10, TGF-β has been implicated in Breg function. B cells have also been implicated in the suppression of allergic airway disease and induction of inhalational tolerance through elaboration of TGF-β [39]. As in our EMT6 murine tumor model, IL-10 did not appear to figure prominently in induction of inhalational tolerance. Another mechanism of inhibition of immune response has also been described involving secretion of IgG linked to LAP-TGF-β. This unique fused form appears to reduce CTL response [40]. This mechanism, only recently described, has not yet been observed in the context of malignancy.
In another tumor model, metastatic disease of the lung following orthotopic implantation of mammary carcinoma cells also appeared to be dependent on TGF-β elaborated by B-regulatory cells. The B-regulatory cell population appears to be evoked by tumor implantation and resembled a subset of B cells called B2 cells (CD19+CD25+CD69+) [41]. These tumor-evoked Bregs were able to support conversion of CD4+ T cells into Tregs, which were FoxP3+. Interestingly, in the absence of tumor-evoked Breg, these investigators did not see metastatic disease into the lung in part due to lack of Tregs [41]. Recently, other investigators have demonstrated significant Treg and B cell infiltration into human squamous cell carcinomas of the lung [1]. Whether similar mechanisms are operative relative to those encountered in the aforementioned murine model is not clear. In the EMT6 model, a very small percentage of tumor-infiltrating B cells express CD25, suggesting that B cells may differentiate differently or that an alternative B cell subset is involved in attenuating the immune response.
15.4 B-Regulatory Cell Infiltration into Human Tumors
In contrast to extensive data associating Treg infiltration with immune suppression and prognosis in human tumors, the prevalence and extent of B cell infiltration in human tumors have been poorly characterized to date. Extensive B cell infiltration has been described in ovarian carcinoma and lung cancer, as well as in squamous cell carcinomas of the oropharynx, and correlated to poor outcome [1–3]. Interestingly B cell infiltration into tongue lesions appears to correlate with progression from early hyperkeratosis to frank carcinoma of the oropharynx [2]. In advanced renal cell carcinoma (RCC), B cell infiltrates have been described which appear to be out of proportion to the relative number of B cells circulating in the peripheral blood [42]. B cell infiltration is thought to potentiate a Th2 response, in the case of RCC.
Despite the immune suppressive attributes of tumor-infiltrating B cells, other human tumors such as medullary carcinomas of the breast may carry a good prognosis in relation to B cell infiltration [43, 44]. Reasons behind extensive B cell infiltration in medullary breast carcinoma have not been elucidated, and whether the presence of B cells is critical to ongoing growth of the tumor regardless of prognosis has also not been established.
Recently, Ganesan et al. have described significant infiltration of human non-small cell lung carcinoma (NSLSC) with CD4+CD25+FOXP3+ Tregs [1]. The investigators also described significant infiltration with CD20+CD19+HLA-DR+ B cells. Although no immune suppressive benefit has been attributed to B cell infiltrates, it is tempting to speculate that they may play an immune suppressive role, either directly as seen in murine tumors or through the support and expansion of Tregs.
15.5 Breg Function in Non-Hodgkin Lymphoma
While the role of Bregs has been extensively characterized in autoimmune diseases, the role in malignant disorders is less clear. Since malignant B cells retain many of the underlying characteristics of normal B cells, it is reasonable to assume that they may invoke B-regulatory cell pathways to suppress T cell responses. Malignant B cells may be involved in multiple pathways that inhibit antitumor immune response. To facilitate their own development and avoid immune elimination, lymphoma B cells may target the balance of T cells, recruiting and expanding Tregs while inhibiting killer CD8+ T cells and helper T cells [45, 46]. It is known that antitumor response in some B cell non-Hodgkin lymphoma (NHL) is profoundly suppressed by the presence of large numbers of intratumoral Tregs [47]. Malignant B cells may alter the overall balance of Tregs and so-called Th17 cells [45, 48]. Skewing the balance toward Treg activity may facilitate tumor survival. It appears that a reciprocal regulatory relationship may exist between Tregs and Th17 cell numbers. B7-H1+ Tregs have been known to infiltrate B cell-derived NHL, thereby inhibiting the proliferation of T cells in a B7-H1 (also known as PD-L1 or CD274)-dependent mechanism [48]. Samples from NHL patients were observed to have significantly lower CD4+ IL-17-producing T cells compared with samples from patients with benign hyperplastic lymph nodes. In the absence of lymphoma B cells, treatment with IL1-β/IL-6 or lipopolysaccharide (LPS) enhanced IL-17 expression in CD4+ T cells; nevertheless, this enhancement was attenuated when CD4+ T cells were cocultured with lymphoma B cells [45]. In the presence of lymphoma B cells, Th17 cell generation was inhibited. Conversely, depletion of lymphoma B cells using anti-CD19 antibody resulted in the enhanced generation of IL-17-producing T cells by IL1-β/IL-6. Both IL-1-β and IL-6 delivered alone or in combination increased the number of CD4+ IL-17-producing cells [45].
Lymphoma B cells may also contribute to the expansion of Tregs, thereby leading to the attenuation of CD8+ response [46]. When intratumoral Tregs were cocultured with infiltrating activated CD8+ T cells, CD8+ cytotoxic activity against lymphoma B cells was significantly inhibited when compared to infiltrating activated CD8+ T cells alone. Conversely, cocultures of infiltrating activated CD8+ T cells with CD4+CD25− T effector cells did not have a significant effect on cytotoxic activity when compared to infiltrating activated CD8+ T cells alone [46]. Hence, B cells may skew responses due to their effects on Th17 and Treg generation. This, in turn, may affect levels of CD8+-mediated cytotoxic response. The prognostic significance of Th17/Treg balance remains to be established.
A mechanism by which lymphoma B cells induce Treg expression has recently been described. Stimulated by TGF-β and IL2, CD4+CD25– cells can convert into CD4+CD25+ Tregs and express Foxp3 [49–52]. Non-Hodgkin lymphoma B cells were also found to induce the expression of Foxp3 in CD4+CD25– cells [53]. Additionally, follicular lymphoma B cells have been shown to produce IL-12, which in turn can promote T cell immunoglobulin and mucin protein 3 (TIM-3) expression on intratumoral T cells. TIM-3 has been characterized as a marker of T cell exhaustion and functional impairment. The observation of high levels of serum IL-12 and increased numbers of TIM-3+CD4 and TIM3+CD8+ T cells has been correlated to worse outcome in patients with follicular B cell NHL [54].
In conclusion, malignant lymphoma B cells may actively support the development of Tregs and may also inhibit Th17 generation. Lymphoma B cells have also been demonstrated to facilitate the production of IL-12, which can in turn lead to T cell exhaustion as identified by TIM-3 expression. These results require confirmation but clearly suggest a direct role for malignant B cells in suppression of antitumor response in man.
15.6 Effects of Depletion of B Cells on Antitumor Immunity
Selective depletion of B cells has been employed as a therapeutic maneuver in the context of both autoimmune disease and lymphoma. The chimeric anti-CD20 monoclonal antibody (mAb) rituximab was the first of several antibodies directed at CD20 antigen associated with B cells approved for human use [55]. Rituximab appears to mediate depletion of normal and memory B cells in patients with autoimmune disease, as well as malignant B cells in the context of lymphoma. The mechanism of action of rituximab is thought to depend primarily upon antibody-dependent cellular cytotoxicity (ADCC); however, under conditions of high antibody density, rituximab can fix complements as well and mediate complement-dependent cytotoxicity (CDC). Rituximab is also thought to engender delayed T cell responses, and this property is felt to relate to Fc receptor-mediated immunization [56]. B cell depletion using rituximab has been successfully employed in a variety of autoimmune diseases. Interestingly in some cases, depletion of pathogenic B cell populations has been shown to be effective in the setting of autoimmune disease such as SLE or rheumatoid arthritis [57–59]. To understand this observation it is important to note that some CD20– B cell subsets have been implicated as possessing B-regulatory cell activity and may be enriched following rituximab depletion of CD20+ B cells [60]. It is also conceivable that various B cell subsets may be affected differently by rituximab-mediated B cell depletion and that this might result in variable effects. In animal models, B cell depletion using anti-CD20 antibodies is only partially effective, with less than complete depletion of B cells observed in the spleen and bone marrow compared to blood and lymph nodes [61].
Related posts:
 Novel Strategy of Cancer Immunotherapy: Spiraling Up
Novel Strategy of Cancer Immunotherapy: Spiraling Up
 Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
 Polarization of Tumor Milieu: Therapeutic Implications
Polarization of Tumor Milieu: Therapeutic Implications
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response
 Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


