- 5.1 Definition of autoimmunity and autoimmune disease
- 5.2 Patterns of autoimmune disease
- 5.2.1 Organ-specific autoimmune disease
- 5.2.2 Non-organ-specific autoimmune disease
- 5.2.3 IgG 4-related disease
- 5.2.1 Organ-specific autoimmune disease
- 5.3 Who gets autoimmune disease?
- 5.4 What prevents autoimmunity?
- 5.4.1 Autoimmunity and self-tolerance
- 5.4.2 Thymic tolerance
- 5.4.3 Peripheral tolerance
- 5.4.4 B-cell tolerance
- 5.4.1 Autoimmunity and self-tolerance
- 5.5 How does tolerance break down?
- 5.6 What triggers autoimmunity?
- 5.6.1 Genetic factors
- 5.6.2 Environmental factors
- 5.6.1 Genetic factors
- 5.7 Mechanisms of tissue damage
- 5.8 Treatment of autoimmune disease
 Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
5.1 Definition of autoimmunity and autoimmune disease
Autoimmunity is an immune response against a self-antigen or antigens. Autoimmune disease is tissue damage or disturbed physiological function due to an autoimmune response. This distinction is important, as autoimmune responses can occur without disease (Case 5.1) or result from diseases caused by other mechanisms (such as infection). Proof that autoimmunity causes a particular disease requires a number of criteria to be met, as in Koch’s postulates for microorganisms in infectious diseases (Table 5.1). The best evidence for autoimmunity in human disease comes from active transfer of IgG autoantibodies across the placenta in the last trimester of pregnancy, which may lead to the development of transient autoimmune disease in the fetus and neonate (Table 5.2 and Case 5.2). In contrast, there are diseases that are reliably associated with both T-cell and B-cell autoimmune responses but where the precise mechanism of disease is unclear. For example, the chronic liver disease primary biliary cirrhosis (section 14.8.2) is strongly associated with autoantibodies directed against mitochondria (and more specifically against a single isoform of the mitochondrial enzyme pyruvate dehydrogenase), but it seems unlikely that these antibodies play any role in liver damage but are produced secondarily to the damage of unknown aetiology. In another autoimmune disease, myasthenia gravis, transfer of the primary or pathogenic autoantibody causes myasthenic features in a rabbit. Caution is therefore required, both in making the assumption that autoimmune responses necessarily imply autoimmune disease and that a given autoimmune antibody or T cell plays a role in the pathogenesis.
 Case 5.1 Is this rheumatoid arthritis?
Case 5.1 Is this rheumatoid arthritis?A 43-year-old woman presented to her general practitioner with sudden onset of acute back pain while gardening, followed by more sustained but less severe pain over the next 2 weeks. The GP felt that this was mechanical back pain but performed some ‘screening investigations’ that included a normal CRP level and a positive test for rheumatoid factor at a titre of 1 in 256. She was then referred to her local rheumatology department with a possible diagnosis of “rheumatoid arthritis”. This caused the patient considerable anxiety as her aunt had had severe rheumatoid arthritis, leading to a very high level of disability. When she was seen in the rheumatology clinic 3 months later she still had minor back pain, but this was overshadowed by her anxiety. She had no other musculoskeletal symptoms and examination was normal apart from mild restriction of the lumbar spine. The rheumatologist agreed with the initial diagnosis of mechanical back pain and explained that around 5% of healthy normal people have a positive test for rheumatoid factor. The presence of a normal CRP level was reassuring and testing for rheumatoid factor not useful; it is only used in patients with a clinical diagnosis of rheumatoid arthritis, when it is an helpful prognostic indicator.
Table 5.1 Criteria that confirm that a particular autoimmune response causes a corresponding autoimmune disease
| Criterion | Comment |
|---|---|
| 1 Autoantibodies or autoreactive T cells with specificity for the affected organ are found reliably in the disease | This criterion is met in most endocrine autoimmune diseases. It is more difficult to fulfil where the target antigen (if any) is unknown, as in rheumatoid arthritis Autoantibodies are much easier to detect than autoreactive T cells but autoantibodies can also be detected in some healthy subjects |
| 2 Autoantibodies and/or T cells are found at the site of tissue damage | True for some haematological and endocrine diseases, SLE and some forms of glomerulonephritis and muscle or muscle end-plate diseases |
| 3 The levels of autoantibody or T-cell response reflect disease activity | True for some diseases; demonstrable in acute systemic autoimmune diseases with rapidly progressive tissue damage, such as some subjects with renal SLE, systemic vasculitis or antiglomerular basement membrane disease |
| 4 Reduction of the autoimmune response leads to improvement | Benefits of immunosuppression are seen in many disorders, but most immunosuppressive treatments are non-specific and anti-inflammatory Furthermore autoantibodies have a half-life of 3–4 weeks so reductions in titres are slower than clinical recovery |
| 5 Transfer of antibody or T cells to a second host leads to development of autoimmune disease in the recipient | Easily demonstrated in animal models, such as myasthenia. In humans, by transplacental transfer of autoreactive IgG antibodies during the last third of pregnancy (see Case 5.2) and by development of autoimmune disease in the recipient of bone marrow transplants when the donor has an autoimmune disease |
| 6 Immunization with autoantigen and consequent induction of an autoimmune response causes disease | Many self-proteins induce an autoimmune response in animals but only if injected with an appropriate adjuvant. Harder to demonstrate in humans, but in the past rabies immunization involved use of infected (but non-infective) mammalian brain tissue, which induced autoimmune encephalomyelitis |
SLE, Systemic lupus erythematosus.
Table 5.2 IgG antibody-mediated diseases capable of placental transfer
| Maternal autoantibody to | Disease induced in neonate |
|---|---|
| Thyroid-stimulating hormone | Neonatal Graves’ disease |
| Epidermal basement membrane cell adhesion molecules | Neonatal pemphigoid |
| Red blood cells | Haemolytic anaemia |
| Platelets | Thrombocytopenia |
| Acetylcholine receptor | Neonatal myasthenia gravis |
| Ro and La | Neonatal cutaneous lupus and congenital complete heart block |
 Case 5.2 Myasthenia gravis and neonatal myasthenia gravis
Case 5.2 Myasthenia gravis and neonatal myasthenia gravisA 21-year-old woman was referred to a neurology clinic with a 1-month history of double vision, difficulty swallowing and weakness in her upper arms. These symptoms were mild or absent in the morning and tended to worsen through the day. When she was seen towards the end of an afternoon neurology clinic, she was found to have a bilateral ptosis and disconjugate eye movements that could not be ascribed to a cranial nerve lesion. Her upper limb power was initially normal but deteriorated with repeated testing. An intravenous injection of edrophonium, a short-acting cholinesterase inhibitor, completely abolished the neurological signs but her eye movements deteriorated again 30 min after the injection. A clinical diagnosis was made of myasthenia gravis. Subsequent blood testing showed the presence of a high level of autoantibodies against the acetylcholine receptor.
She was treated with oral cholinesterase inhibitors with some improvement. However, 1 month later she deteriorated and corticosteroids were introduced without effect. A computed tomography scan of her thorax showed no evidence of a thymoma but she was nevertheless referred to a thoracic surgeon for thymectomy, as this can sometimes induce remission in myasthenia even in the absence of a thymoma. A small thymic remnant was removed and she recovered uneventfully and was able to withdraw from all medication without deterioration in her symptoms. Acetylcholine receptor antibody levels fell but remained detectable. One year later, she became pregnant and after an uneventful 41-week pregnancy she delivered a 4-kg male infant. There were immediate concerns about the baby, who failed to make adequate respiratory efforts and who appeared limp and hypotonic. The baby was intubated and ventilated on the neonatal intensive care unit. In view of mother’s history, a provisional diagnosis of neonatal myasthenia gravis was made, although care was taken to exclude other causes of neonatal respiratory insufficiency such as maternal analgesia with pethidine, hypoglycaemia and sepsis. A cranial ultrasound showed no evidence of bleeding or other pathology. Subsequent testing of a blood sample taken from the umbilical cord showed low levels of acetylcholine receptor antibody. The baby needed ventilation and feeding via a nasogastric tube for 3 days, after which time the ventilation was successfully withdrawn. There were some initial feeding problems due to difficulty sucking and swallowing, but these resolved over the next 48 h. The child’s subsequent development has been entirely normal. The mother also remains well.
5.2 Patterns of autoimmune disease
Autoimmune diseases can affect any organ in the body, although certain systems seem particularly susceptible (e.g. endocrine glands). Autoimmune diseases have been conventionally classified into (i) organ-specific and (ii) non-organ-specific disorders; though this is an arbitrary division, it is useful in thinking about the pathogenesis of each condition.
5.2.1 Organ-specific autoimmune disease
Organ-specific autoimmune disorders (Cases 5.2 and 5.3) usually affect a single organ and the autoimmune response is directed against multiple antigens within that organ. Most of the common organ-specific disorders affect one or another endocrine gland. The antigenic targets may be molecules expressed on the surface of cells (particularly hormone receptors) or intracellular molecules, particularly intracellular enzymes (Table 5.3).
 Case 5.3 Fungal infections, fits and hypocalcaemia
Case 5.3 Fungal infections, fits and hypocalcaemiaA 14-year-old boy presented to a dermatologist with sore, cracked hypertrophic lips, chronic paronychia (tender, swollen nail beds with dystrophic nails) and curious horn-like lesions in the scalp. The dermatologist made a clinical diagnosis of chronic mucocutaneous candidiasis, and subsequently cultured the yeast, Candida albicans, from the boy’s mouth and a dermatophyte fungus from the lesions on the scalp. The dermatologist noted a history of epilepsy starting at the age of 5. Subsequent investigation demonstrated profound hypocalcaemia with corrected serum calcium of 1.1 mm/l (normal 2.2–2.6) with undetectable levels of parathyroid hormone. His 4-year-old sister had also recently developed epilepsy and was also found to be severely hypocalcaemic. The classical clinical picture allowed a confident diagnosis of autoimmune hypoparathyroidism (subsequently confirmed by positive autoantibodies against endocrine parathyroid tissue) as a feature of APECED (Autoimmune Polyendocrinopathy, Candidiasis and Ectodermal Dysplasia). The patient subsequently developed fatigue and vomiting and a short synacthen test revealed adrenal cortical failure as well and he was found to have autoantibodies to adrenal cortex confirming autoimmune adrenalitis. As yet there is no evidence of diabetes mellitus. Genetic analysis confirmed a disease causing mutation in the AIRE gene and the other family members were also screened.
Table 5.3 Some examples of self-antigens and associated diseases. More information can be found in the appropriate organ-based chapters. In general, tissue-specific antigens are associated with organ-specific diseases and those antigens found in all cells are associated with systemic disease
| Self-antigen | Disease |
|---|---|
| Hormone receptors | |
| TSH receptor | Hyper- or hypothyroidism |
| Insulin receptor | Hyper- or hypoglycaemia |
| Neurotransmitter receptor | |
| Acetylcholine receptor | Myasthenia gravis |
| Cell adhesion molecules | |
| Epidermal cell adhesion molecules | Blistering skin diseases |
| Plasma proteins | |
| Factor VIII | Acquired haemophilia |
| β1-glycoprotein I and other anticoagulant proteins | Antiphospholipid syndrome |
| Other cell-surface antigens | |
| Red blood cells (multiple antigens) | Haemolytic anaemia |
| Platelets | Thrombocytopenic purpura |
| Intracellular enzymes | |
| Thyroid peroxidase | Hypothyroidism |
| Steroid 21-hydroxylase (adrenal cortex) | Adrenocortical failure (Addison’s disease) |
| Glutamate decarboxylase (β-cells of pancreatic islets) | Autoimmune diabetes |
| Lysosomal enzymes (phagocytic cells) | Systemic vasculitis |
| Mitochondrial enzymes (particularly pyruvate dehydrogenase) | Primary biliary cirrhosis |
| Intracellular molecules involved in transcription and translation | |
| Double-stranded DNA | SLE |
| Histones | SLE |
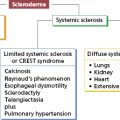
| Topoisomerase I | Diffuse scleroderma |
| Amino-acyl t-RNA synthases | Polymyositis |
| Centromere proteins | Limited scleroderma |
TSH, Thyroid-stimulating hormone; SLE, systemic lupus erythematosus.
 Visit the website at www.immunologyclinic.com to read case studies on primary autoimmune hypothyroidism, and IL-2 treatment in rheumatoid arthritis.
Visit the website at www.immunologyclinic.com to read case studies on primary autoimmune hypothyroidism, and IL-2 treatment in rheumatoid arthritis.
5.2.2 Non-organ-specific autoimmune disease
Non-organ-specific disorders affect multiple organs and are usually associated with autoimmune responses against self-molecules that are widely distributed through the body, and particularly with intracellular molecules involved in transcription and translation of the genetic code (Table 5.3). Many of these non-organ-specific disorders fall within the group of multisystem disorders labelled as ‘connective tissue diseases’; however, this is a misleading term since the ‘connective tissues’ are often neither abnormal nor specifically damaged, but the term is still in widespread use unfortunately.
5.2.3 IgG 4-related disease
A recently described condition, in which both lymphoproliferation and autoimmunity are significant, is the oddly named ‘ImmuoglobulinG4-related disease (IgG4-RD)’. Originally recognized as autoimmune pancreatitis (acute abdominal pain and gross lymphadenopathy) particularly in middle-aged men in Japan, this syndrome has much wider organ involvement affecting nearly every organ system, such as the gastrointestinal tract, including the salivary glands, and also lymph nodes, retroperitoneum, blood vessels and many other organs. The only common findings are raised serum immunoglobulins due to excessive levels of IgG4, positive non-specific autoantibodies (rheumatoid factor, antinuclear antibodies) and variable increases in serum lactate dehydrogenase levels. Since serum levels of IgG4 are normally extremely low, and many autoantibodies are known to be of IgG 4 isotype, this condition is thought to be largely autoimmune in nature. On biopsy, the histology in all tissues is remarkably similar: diffuse plasmacytic infiltrate of IgG4+ plasma cells with variable fibrosis and sometimes with reactive lymphoid follicles which can resemble lymphoma. Unlike the other IgG subclass molecules, the heavy chains of the IgG4 molecule are unstable and 50% of IgG4 molecules consist of heavy chains linked weakly by non-covalent forces. This means that the heavy chains can separate and recombine randomly, making new antigen-binding sites but failing to form immune complexes for clearance of antigen. Whether or not this is important in the pathogenesis of this variable condition remains to be seen. Patients have not yet been followed long enough to know whether this is a pre-malignant condition and the long-term outlook is not yet known. Treatment consists of corticosteroids (usually very effective) and Rituximab in resistant or recurring disease.
5.3 Who gets autoimmune disease?
The burden of autoimmune diseases is considerable throughout the world; around 3% of the population has an autoimmune disease. Many of the major chronic disabling diseases affecting people of working age are considered to have an autoimmune basis. These include multiple sclerosis (MS), rheumatoid arthritis (RA) and insulin-dependent diabetes mellitus (IDDM). Autoimmune diseases are rare in childhood and the peak years of onset lie between puberty and retirement age, the major exception being the childhood-onset form of diabetes mellitus.
There are striking gender differences in the risk of developing an autoimmune disease. Almost all are more common in women, and for some autoimmune diseases the risk is increased eight times in females. There are, however, notable exceptions, such as ankylosing spondylitis, which is rare in women.
The prevalence of autoimmunity tends to be higher in northern latitudes, is probably higher in industrialized societies, and seems to increase progressively as the pattern of social and economic organization develops. It is unclear whether this geographic and socioeconomic variation in autoimmunity reflects differential exposure to pathogens, variations in nutrition, ascertainment of disease or other factors.
Autoimmune diseases also show evidence of clustering within families (Case 5.3, Table 5.4) and most show polygenic features that are slowly being unravelled with next-generation sequencing of exomes or genomes. These genetic factors are discussed in more detail in section 5.6.1.
Table 5.4 Single gene defects that provide an insight into autoimmune diseases
| Gene defect or experimental genetic manipulation | Autoimmune disease seen as consequence of gene defect | Implications for autoimmunity |
|---|---|---|
| Deficiency of early classical complement pathway components, C1q, C2, C4 in humans and knockout mice Defects in autophagy genes in humans and knockout mice | Systemic lupus erythematosus | Early classical complement pathway important in immune-complex clearance and autophagy provides recycling of cell components and disposal of intracellular debris |
| Chronic granulomatous disease (defect in NADPH oxidase enzyme complex) in human female carriers | Discoid lupus erythematosus | Phagocytes scavenge and destroy cell debris preventing activation by self antigens |