At the conclusion of this chapter, the reader should be able to: • Describe the nature of autoimmune disorders. • Compare organ-specific and organ-nonspecific characteristics. • Describe organ-specific and midspectrum disorders. • Analyze representative case studies. • Correctly answer case study related multiple choice questions. • Be prepared to participate in a discussion of case study related critical thinking questions. • Describe the principle, sources of error, limitations, and application of the antinucleoprotein slide test. The major autoimmune diseases, e.g., systemic lupus erythematosus (see Chapter 29), rheumatoid arthritis (see Chapter 30), diabetes type 1, and multiple sclerosis share many common features. Chronic and other intermittent inflammation contributes over time to the destruction of target organs that contain inciting antigens or are the sites of immune-complex deposition. Although the adaptive immune system has long been the focus of attention, innate immune mechanisms are now viewed as central to the pathogenesis of these disorders. New genetic findings emphasize the identification of environmental components that interact with host genetic factors are being important to developing a deeper understanding of autoimmunity. The term autoimmune disorder is used when demonstrable immunoglobulins (autoantibodies) or cytotoxic T cells display specificity for self antigens, or autoantigens, and contribute to the pathogenesis of the disorder (Table 28-1). Autoimmune disorders are characterized by the persistent activation of immunologic effector mechanisms that alter the function and integrity of individual cells and organs. The sites of organ or tissue damage depend on the location of the immune reaction. The variety of signs and symptoms seen in patients with autoimmune disorders reflects the various forms of the immune response. Table 28-1 Autoimmune Disorders and Associated Abnormalities Many disorders are believed to be related to immunologic abnormalities and additional diseases are continually being identified (Box 28-1). Autoimmune disorders exhibit a full spectrum of tissue reactivity (Fig. 28-1). At one extreme are organ-specific disorders such as Hashimoto’s disease of the thyroid; at the other extreme are disorders that manifest as organ-nonspecific diseases, such as systemic lupus erythematosus (SLE; see Chapter 29) and rheumatoid arthritis (RA; see Chapter 30; Table 28-2). Table 28-2 Summary of Organ-Specific and Organ-Nonspecific Disorders An individual may develop an autoimmune response to a variety of immunogenic stimuli (Table 28-3). These responses may be caused by the following: Table 28-3 Antigens Implicated in Autoimmune Endocrine Diseases • Antigens that do not normally circulate in the blood The hidden antigen (sequestered antigen) theory is one of the earliest views of organ-specific antibodies. Antigens are sequestered within the organ and, because of the lack of contact with the mononuclear phagocyte system, they fail to establish immunologic tolerance. Any conditions producing a release of antigen would then provide an opportunity for autoantibody formation. This situation occurs when sperm cells or lens and heart tissues are released directly into the circulation, and autoantibodies are formed. Unmodified extracts of tissues involved in organ-specific autoimmune disorders, however, do not readily elicit antibody formation. • Altered antigens that arise because of chemical, physical, or biological processes (e.g., hapten complexing, physical denaturation, mutation) • A foreign antigen that is shared or cross-reactive with self antigens or tissue components • Mutation of immunocompetent cells to acquire a response to self antigens • Loss of the immunoregulatory function by T lymphocyte subsets • Elimination of the small clone of immunocompetent cells programmed to react with the antigen (Burnet’s clonal selection theory) • Induction of unresponsiveness in the immunocompetent cells through excessive antigen binding to them and triggering of a suppressor mechanism Major autoantibodies can be detected in different disorders. Many diagnostic laboratory tests (Box 28-2) are based on detecting these autoimmune responses. Common autoantibodies include thyroid, gastric, adrenocortical, striated muscle, acetylcholine receptor, smooth muscle, salivary gland, mitochondrial, reticulin, myelin, islet cell, and skin. Antibodies to antinuclear antibodies (ANAs) include deoxyribonucleic acid (DNA), histone, and nonhistone protein antibodies. Vasculitis is characterized by inflammation within blood vessels, which often results in a compromise of the vessel lumen with ischemia. Ischemia causes the major manifestations of the vasculitic syndromes and determines the prognosis. Any size and type of blood vessel may be involved. Therefore, the vasculitic syndromes are a heterogeneous group of diseases (Box 28-3). Lymphoid thyroiditis is believed to be the most common cause of sporadic goiter. Characteristically, there is a firm, diffusely enlarged, nontender thyroid gland that may be lobulated. Hypothyroidism, however, is a common late sequela of lymphoid thyroiditis, and patients are usually euthyroid when first seen by a physician. Some individuals have clinical and pathologic evidence of the coexistence of Graves’ disease and lymphoid (Hashimoto’s) thyroiditis. Histologically, Hashimoto’s thyroiditis is characterized by diffuse lymphocytic infiltration (Fig. 28-2). Antibodies directed against thyroid microsomal antigen (thyroid peroxidase antibody [anti-TPO]) can be detected by various techniques (Table 28-4). Chemiluminescent immunoassay is typically performed to detect anti-TPO autoantibodies. TPO plays a significant role in the biosynthesis of thyroid hormones by catalyzing the iodination of tyrosyl residues in thyroglobulin and the coupling of iodotyrosyl residues to form T4 and T3. Autoantibodies produced against TPO are capable of inhibiting enzyme activity. They are also complement-fixing antibodies that can induce cytotoxic changes in cells and consequently cause thyroid dysfunction. More than 90% of patients with autoimmune thyroiditis (Hashimoto’s thyroiditis) have anti-TPO. Antibodies to TPO have also been found in most patients with idiopathic hypothyroidism (85%) and Graves’ disease (50%). Table 28-4 T cells of the CD4+ type are responsible for initiating the immune response to the islets that results in islet cell autoantibodies and B cell destruction. Patients with T1D have the following types of autoantibodies (Box 28-4): IA-2 is directed against a phosphatase-type transmembrane 37-kDa islet beta cell antigen (ICA512). • Hypergammaglobulinemia (elevated serum IgG or gamma globulin level) in patients with enhanced peripheral rim halo of the pancreas on CT • Elevated serum IgG4 concentrations in patients with a diffusely enlarged pancreas • Autoantibodies against carbonic anhydrase II (ACA II), lactoferrin (antilactoferrin antibody [ALA]), anti–smooth muscle antibody (ASMA), or ANA • Increased number of CD4+ T lymphocytes in peripheral blood
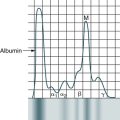
Autoimmune Disorders
What is autoimmunity?
Clinical Diagnosis
Autoantigen
Addison’s disease
P-450 enzymes
Crohn’s disease
p-ANCA, pancreatic acinar cells
Ovarian failure/infertility
P-450 enzymes
Pernicious anemia
Parietal cells
Ulcerative colitis
p-ANCA
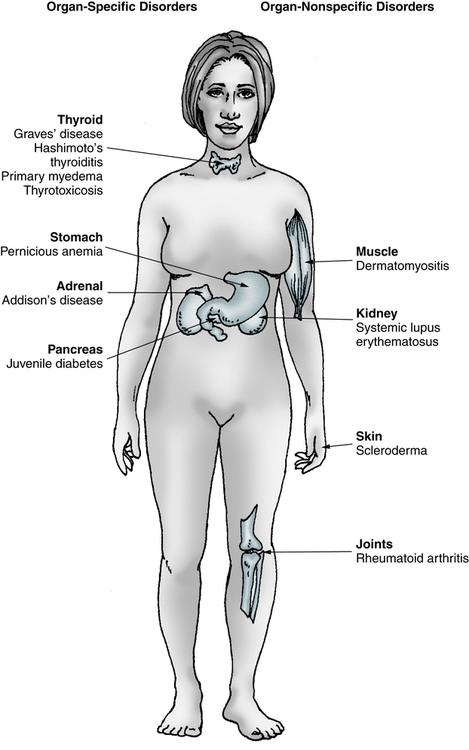
Spectrum of Autoimmune Disorders
Similarities
Differences
Organ-Specific
Organ-Nonspecific
Antibodies and lesions are organ-specific.
Antibodies and lesions are organ nonspecific.
Clinical and serologic overlap (e.g., thyroid, stomach, adrenal glands, kidney).
Overlap of SLE, RA, and other connective tissue disorders.
Antigens only available to lymphoid system in low concentrations.
Antigens accessible at higher concentrations.
Antigens evoke organ-specific antibodies in normal animals with complete Freund’s adjuvant.
No antibodies produced in animals with comparable stimulation.
Familial tendency to develop organ-specific autoimmunity.
Familial tendency to develop connective tissue disease.
Questionable abnormalities in immunoglobulin synthesis in relatives.
Lymphoid invasion, parenchymal destruction by questionable cell-mediated hypersensitivity or antibodies.
Lesions caused by deposition of antigen-antibody (immune) complexes.
Tendency to develop cancer in the organ.
Tendency to develop lymphoreticular neoplasia.

Immunopathogenic Mechanisms
Disorder
Antigen
Hashimoto’s disease
Thyroglobulin
Thyroid peroxidase
Thyrotropin receptor
Graves’ disease
Thyrotropin receptor
Thyroid peroxidase
Thyroglobulin
64-kDa antigen
70-kDa heat shock protein
Type 1 diabetes
Insulin/proinsulin
Insulin receptor
Glutamic acid decarboxylase
B cell release granule
Pancreatic cytokeratin
64-kDa antigen
Glucagon
65-kDa heat shock protein
Addison’s disease
Adrenal cortical cells
55-kDa microsomal antigen
Idiopathic hypoparathyroidism
130- and 200-kDa antigens
Endothelial antigen
Mitochondrial antigen
Self-Recognition (Tolerance)
Major Autoantibodies
Organ-Specific and Midspectrum Disorders
Cardiovascular Disorders
Vasculitis
Endocrine Gland Disorders: Thyroid Disease
Lymphoid (Hashimoto’s) Chronic Thyroiditis
Signs and Symptoms
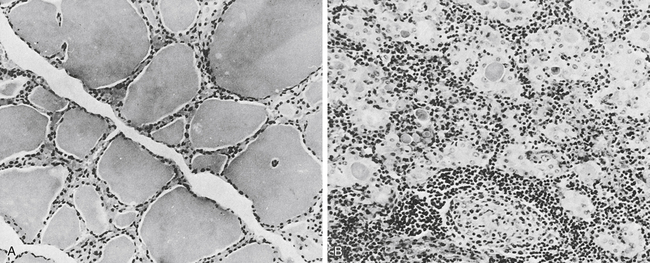
A, In the normal thyroid, colloid fills the vesicles, but in a diseased gland (B), only isolated deposits of colloid are seen. The cell infiltrate is lymphoid in nature. Note germinal center in lower middle (B). (From Anderson JR, Buchanan WW, Goudie RB: Autoimmunity, Springfield, Ill, 1967, Charles C Thomas.)
Diagnostic Evaluation
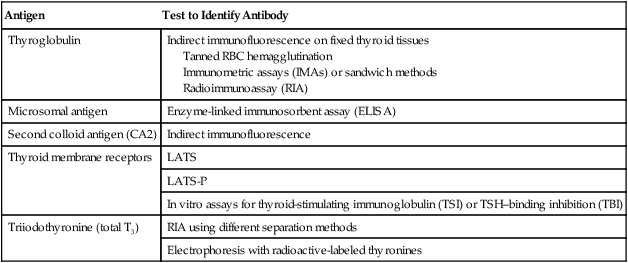
Antigen
Test to Identify Antibody
Thyroglobulin
Indirect immunofluorescence on fixed thyroid tissues
Tanned RBC hemagglutination
Immunometric assays (IMAs) or sandwich methods
Radioimmunoassay (RIA)
Microsomal antigen
Enzyme-linked immunosorbent assay (ELISA)
Second colloid antigen (CA2)
Indirect immunofluorescence
Thyroid membrane receptors
LATS
LATS-P
In vitro assays for thyroid-stimulating immunoglobulin (TSI) or TSH–binding inhibition (TBI)
Triiodothyronine (total T3)
RIA using different separation methods
Electrophoresis with radioactive-labeled thyronines
Pancreatic Disorders
Insulin-Dependent Diabetes Mellitus
Immunologic Manifestations
Autoimmune Pancreatitis
Immunologic Manifestations
Autoimmune Disorders