Gene or marker
Population
Number of patients
Odds ratio
P value
MHC loci
HLA-B*08
Norway [66]
414
2.6
4 × 10−11
HLA-DR3-DQ2
Norway [67]
94
3.6
<10−7
HLA-DR3-DQ2
Finland, Russia, Estonia [68]
69
14.5
<0.0001
HLA-DR4-DQ8*0404
Norway [67]
94
NR
<10−5
HLA-DR4-DQ8*0401
<10−4
HLA-DRB1*0301
Norway [66]
414
22.13
6 × 10−20
HLA-DRB1*0404
Other loci
MICA*5.1
Italy [69]
28
6.52
0.0015
USA [70]
46
22.5
<10−5
Norway [66]
414
1.78
0.0003
CYP21Adel
Finland [71]
12
25.0
NR
CYP21 L10, R102, A494
Finland [72]
12
8.9
NR
CLEC16A (rs12917716)
Norway [73]
332
0.72
0.0004
UK [73]
210
1.06
0.71
Combined [73]
542
0.81
0.006
CYP27B1-126C > A (rs4646536)
Germany [74]
124
1.18
0.006 (genotype)0.3354 (allele)
UK [75]
104
1.71
0.003
6 European cohorts [76]
1955
0.9
0.03
VDR Fok1 (exon2)
Germany [77]
95
NR
0.0351 (genotype)
0.3390 (allele)
FCRL3 (rs7528684)
UK [78]
200
1.61
0.0001
PTPN22 (rs2476601)
UK [79]
104
1.69
0.031
Germa ny [80]
121
1.03
0.9878
Norway [81]
302
1.39
0.016
UK [82]
251
1.63
0.008
Poland [82]
87
1.84
0.010
CTLA4 A > G (exon 1)
UK [83]
90
1.64
0.008
Norway, UK, Germany, Spain and Italy [84]
1002
1.37
0.002
CTLA4 J030G > A
UK [85]
40
1.9
0.02
Norway [85]
94
1.4
0.04
Combined [85]
134
1.5
0.03
NLRP1 (rs12150220)
Norway [86]
333
1.25
0.007
Poland [87]
101
1.5
0.015
PD-L1 (rs1411262)
UK [88]
315
1.33
0.032
Norway [88]
342
1.34
0.026
Combined [88]
657
1.32
3.03 × 10−3
STAT4 (rs4274624)
(rs10931481)
6 European cohorts [76]
1955
1.27
<0.0001
1262
1.23
0.0007
GATA3
6 European cohorts [76]
1955
0.9
0.03
NFκB1 (rs10026278)
(rs230532)
(rs4698861)
UK [76]
309
0.69
0.0034
0.65
0.00041
0.63
0.00017
Il-23A
Italy [76]
280
2.37
0.0028
GPR174 (rs3827440)
UK [89]
286
1.34
0.03
Il-2 (rs3136534)
Poland [90]
223
0.71
0.003
BACH2 (rs3757247)
UK [91]
358
1.44
1.4 × 10−6
Norway
317
1.41
0.0015
NFATC1 (rs754093)
Norway [92]
384
NR
0.48
Sweden [92]
367
NR
0.033 (genotype)
0.07 (allele)
UK [92]
346
NR
0.15
Combined [92]
1097
NR
0.02
Autoimmunity is thought to arise as a result of aberrant responses within both the adaptive and innate immune systems. The adaptive immune system refers to antigen-specific immune responses involving antigen recognition and processing, forming “immune memory”, and concerns its key cells—lymphocytes. Innate immunity refers to non-specific defence mechanisms against pathogens occurring within hours of an antigen’s appearance and comprises a number of cell types. Amongst the functions of the innate immune system are pathogen recognition, production of cytokines and chemokines leading to immune cell recruitment, complement cascade activation and activation of the adaptive immune system via antigen presentation. In patients with AAD, similarly to other autoimmune diseases, aberrant responses in both innate and adaptive immunity are implicated. However, the majority of susceptibility loci identified to date encode proteins involved in antigen presentation and T-cell activation.
MHC Risk Alleles
The major histocompatibility complex (MHC) in humans, the human leukocyte antigen (HLA) complex, is located on chromosome 6p21 and comprises multiple genes involved in immune processes. Amongst those are HLA class I (HLA-A, HLA-B and HLA-C) and HLA class II (HLA-DRB1, HLA-DQB1, HLA-DQA1, HLA-DPB1 and HLA-DPA1) genes which encode antigen-presenting molecules and are the most important determinants of polygenic autoimmune disease risk. HLA proteins are expressed on the surface of antigen-presenting cells and display peptides (both self and non-self) for recognition by T cells. Aberrant activation of T cells in response to self-antigens leads to development of autoimmune disease (Fig. 4.1). Although the exact immunopathology of AAD remains to be established, it has been shown that MHC II molecule expression on adrenocortical cells is highly upregulated in the active phase of the disease [93].
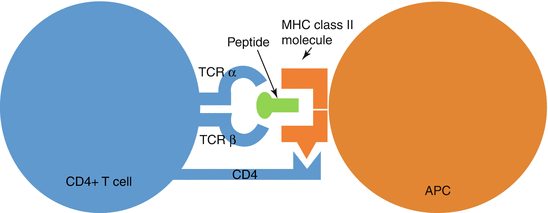
Fig. 4.1
Schematic representation of antigen presentation by MHC class II molecules to CD4+ T cell. The antigen-MHC complex is recognised by the T-cell receptor. CD4 is a co-receptor that binds to a non-polymorphic region of the MHC and assists in T-cell activation. APC, antigen-presenting cell; TCR, T-cell receptor
Similar to other autoimmune conditions, the challenge in finding causal variants within the MHC for AAD lies in the fact that this region contains the largest number of polymorphisms in the entire genome [94] and that there is strong and extensive LD amongst alleles throughout this locus [95]. A number of variants within the MHC class II genes are associated with several autoimmune conditions. In particular, a strong association between autoimmunity and allelic variability in HLA-DR and DQ molecules, which present exogenous antigens for recognition by CD4+ helper (Th) cells, exists. Allelic polymorphism at these loci results in variant proteins that allow self-peptides to enter the antigen-binding groove more readily. The association between HLA class II molecules and AAD has been recognised for three decades [96]: in a seminal study by Maclaren and co-workers, susceptibility to AAD was linked to HLA-DR3 and HLA-DR4 alleles. Subsequently, these findings were replicated in a number of AAD cohorts [66, 67, 71, 97, 98]. A particularly high-risk genotype for AAD has been identified as DR3-DQ2/DR4-DQ8 [67]. To date, the HLA class II alleles DRB1*0301 and DRB1*0404 have been shown to confer the highest risk for AAD with odds ratios (OR) of 2.9 and 3.3, respectively [66]. An especially large risk increment occurs in compound heterozygotes possessing both these haplotypes (OR = 22). The DRB1 alleles occur in strong linkage disequilibrium (LD) with other alleles associated with AAD: DQB1*02 and DQB1*0302 (OR 1.8 and 1.5, respectively) [66]. In addition, a number of haplotypes conferring protection from AAD have been also discovered: DRB1*0401-DQ8 [67] and DRB1*0403 [99]. Thus far, none of the susceptibility loci identified are specific to AAD with the possible exception of HLA-DRB1*0404. The possible explanation for this is that peptides from 21-OH might bind particularly well and be presented to autoreactive T cells by this HLA class II molecule [35].
Polymorphisms in the CYP21A2 (cytochrome P450, family 21, subfamily A, polypeptide 2) gene, which encodes the 21-OH enzyme and is located within MHC class III region, have been associated with AAD. CYP21A2 is 600 kb away from the HLA-DRB1 locus; therefore, the association of its polymorphisms and AAD has been attributed to long-range LD with MHC class II alleles [72]. A recent study confirmed that no specific variants of CYP21A2 are associated with AAD. Instead, CYP21A2 polymorphisms are in LD with the high-risk haplotype HLA-DRB1 locus and do not contribute to the disease susceptibility independently [100].
The genes that appear to be independently associated with AAD susceptibility include HLA-B (OR 2.6 for HLA-B*08) and MHC class I-related chain A (MICA) (OR 1.8 for MICA*5.1) [66]. Homozygosity for the MICA*5.1 allele in the presence of the high-risk HLA genotype DR3-DQ2/DR4.4-DQ8 confers extreme risk for AAD development [101].
Non-MHC Risk Alleles
The CIITA gene encodes a protein functioning as a HLA class II transactivator, the master control factor for MHC class II expression. Mutations in CIITA result in a severe monogenic immunodeficiency disease known as bare lymphocyte syndrome. Allelic variability in this gene has been associated with conditions such as rheumatoid arthritis [102], systemic lupus erythematosus [103] and coeliac disease [104], amongst others. Polymorphisms in the promoter, as well as in intron 3, of this gene have been reported to be associated with AAD susceptibility [73, 105] Although the mechanism by which these polymorphisms confer disease susceptibility remains unknown, it has been hypothesised that they could influence the levels of tissue selectivity of HLA class II expression (Fig. 4.2).
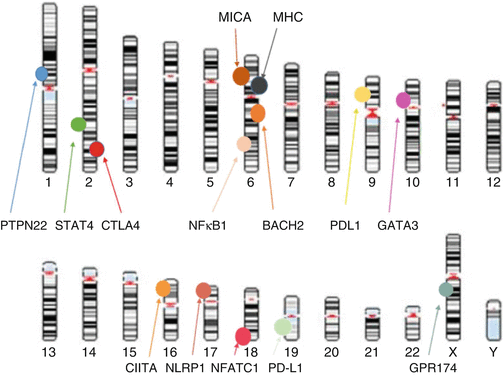
Fig. 4.2
Schematic representation of genes associated with autoimmune Addison’s disease that are implicated in T-cell proliferation and activatio n
T-Cell Proliferation, Differentiation and Activation Genes
Activation of T lymphocytes , the key cells of the adaptive immune system, requires simultaneous engagement of the T-cell receptor by MHC class II peptides and costimulatory molecules. The cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) gene located on chromosome 2q33 encodes CD152, a costimulatory molecule which acts as a vital negative regulator of T-cell activation and proliferation [106]. CTLA4 competes with costimulator CD28 for binding to B7 on antigen-presenting cells. The critical role of this molecule is demonstrated in CTLA4 knockout mice which die prematurely, at an age of 2–3 weeks, due to severe lymphoproliferation, lymphocytic infiltration and destruction of major organs [107]. In humans, mutations in the CTLA4 gene result in an immune dysregulation syndrome [108]. CTLA4 gene polymorphisms have been linked with susceptibility to a number of autoimmune diseases including autoimmune thyroid disease [109, 110], type 1 diabetes mellitus [111, 112], rheumatoid arthritis [113] and coeliac disease [114]. The association between CTLA4 gene variants and AAD has been reported in a number of studies in different European populations [83–85, 115, 116]. The most commonly described CTLA4 polymorphisms in AAD populations are non-synonymous polymorphism in exon 1 region of CTLA4 gene +49 A → G (Ala17Thr) [83, 84, 115], (AT)n dinucleotide repeat polymorphism within the 3′ untranslated region [116] and G or A alleles of the JO30 SNP downstream of this gene [85]. Polymorphisms in the latter are postulated to be the causative variants affecting the relative amount of the soluble and membrane-bound CTLA4 and therefore enabling CD28 to access more of its ligand, resulting in T-cell activation [117]. In contrast to this hypothesis, a number of studies have demonstrated that individuals with autoimmune conditions have, in fact, increased serum levels of soluble CTLA4 isoforms [118, 119] suggesting that the complexity of CTLA4-CD28 interaction and signalling is incompletely understood. Another mechanism of negative immune regulation by CTLA4-positive cells is the ability of CTLA4 to capture and degrade CD80 and CD86 (ligands shared with the stimulatory receptor CD28) from antigen-presenting cells [120].
Another key autoimmunity gene, PTPN22 on chromosome 1p13, encodes a tyrosine phosphatase which is a crucial regulator of immune homeostasis, inhibiting T-cell receptor (TCR) signalling. The association of a missense SNP (1858C > T) in PTPN22, encoding an arginine to tryptophan substitution at amino acid 620, has been identified in type 1 diabetes [121], rheumatoid arthritis [122], systemic lupus erythematosus [123] and Graves’ disease [79]. For a number of autoimmune diseases, this allelic variant ranks as the most important non-MHC single gene contributor to disease susceptibility. A number of stu dies have implicated this variant in susceptibility to AAD [79, 81, 82]. The functional effect of 1858 C > T polymorphism has not been fully elucidated. Some studies suggest that this variant results in increased suppression of TCR signalling [124, 125], while others suggest the opposite [126, 127]. One study suggested that proteolytic binding and cleavage of Arg620Trp is increased with consequent reduction in LYP levels in T and B cells leading to lymphocyte and dendritic cell hyperresponsiveness and autoimmunity [127].
Programmed death ligand-1 (PD-L1, CD274, B7-H1) is a costimulatory molecule binding the PD-1 moiety of T cells, leading to downregulation of cytokine production and T-cell activation, thereby inducing immune tolerance. PD-L1 variants have been implicated in susceptibility to AAD, although the effect on risk is very modest (OR 1.34 for the allele with the strongest association) [88].
Interleukin-2 (Il-2) and its receptor are important determinants of the immune response. Il-2 is a potent T-cell growth stimulator and influences T-cell differentiation, in particular formation of the regulatory T cell (Treg) lineage. Treg cells are crucial in maintaining self-tolerance due to their ability to suppress autoreactive T cells which escape negative selection in the thymus. IL2RA gene encodes the alpha subunit (CD25) of the IL2 receptor, a unique subunit conferring high affinity to IL2. Polymorphic variants of IL2 (4q27) and IL2RA (10p15.1) genes have been associated with type 1 diabetes and rheumatoid arthritis [128–130]. The C minor allele in IL2 conferred protection from AAD in a Polish cohort. However, there was no association found between IL2 polymorphisms and Norwegian patients with AAD [73]. Similarly, an association between IL2RA polymorphisms and susceptibility to AAD was found in Norwegian subjects, but this finding has not been replicated in British or Polish series [73, 90]. Although there is known genetic heterogeneity between various AAD populations, the association between AAD and IL2 or IL2RA needs further replication in larger cohorts.
STAT4 (signal transducer and activator of transcription 4 ) on chromosome 2q32 encodes a transcription factor which is implicated in Th-1 and Th-17 differentiation and activation. Polymorphisms of STAT4 have been shown to be associated with rheumatoid arthritis [131–133], systemic lupus erythematosus [122] and type 1 diabetes [134]. A meta-analysis by Mitchell et al. revealed a significant association between STAT4 polymorphisms and AAD in European populations [76]. The allelic variability identified in AAD maps to intron 3 of the STAT4 gene and is in moderate-to-strong linkage disequilibrium with STAT4 polymorphisms identified in the other autoimmune conditions. The exact mechanism by which polymorphisms in this gene lead to autoimmune disease remains unknown.
The GATA3 gene on chromosome 10p14 encodes a C2C2-type zinc finger transcription factor which regulates a number of steps in T-cell development and differentiation. In particular, this transcription factor has been shown to be the Th2 lineage master regulator [135] and could therefore contribute to T-cell dysregulation present in autoimmune disease. A recent meta-analysis demonstrated an association between GATA3 polymorphisms and AAD in European cohorts [76]. The minor G allele at rs3802604 was protective for AAD (OR 0.9). This finding is, however, in contrast to the known association of the same allele with susceptibility to rheumatoid arthritis [136] possibly reflecting immunopathogenic differences between these two conditions.
The BACH2 gene on chromosome 6q15 plays a vital role in CD4+ T-cell differentiation; in particular it is crucial in the formation of Treg, which are the key cells in maintaining immune tolerance. Polymorphisms at the BACH2 locus have been associated with type 1 diabetes, generalised vitiligo, autoimmune thyroid disease, Crohn’s disease and coeliac disease [137–141]. Recently, an association between the minor T allele at rs3757247 in the BACH2 locus and susceptibility to AAD was described in UK and Norwegian cohorts [91].
The first linkage study in multiplex AAD families implicated regions on chromosomes 6 (corresponding to the HLA region), 7, 9 and 18 in the susceptibility to AAD [92]. A follow-on study, looking at 64 SNPs underlying the linkage peaks on chromosomes 9 and 18 conducted in case-control cohorts from the UK, Norway and Sweden, revealed nominal association with three independent SNPs in chromosome 18 and AAD. One of these encodes a non-synonymous variant (pCys751Gly) in exon 9 of the NFATC1 (nuclear factor of activated T cells, cytoplasmic, calcineurin-dependent 1) gene. Upon T-cell activation, family members of NFAT translocate to the nucleus where they can activate target genes and as such play a central role in gene transcription during the immune response [142]. NFATC1 has been shown to play a role in the regulation of PD-1 expression, a cell surface receptor functioning as an immune checkpoint and reducing T-cell activation [143].
The GPR174 (G protein-coupled receptor 174) gene at Xq21.2 consists of one exon encoding a protein which belongs to the G protein-coupled receptor superfamily and is involved in immune cell signal transduction. Recently a lysophosphatidylserine (LysoPS) was found to be a ligand for GPR174, and the interaction between the two has been s hown to stimulate an increase in the intracellular cyclic adenosine monophosphate (cAMP) [144]. cAMP has been previously demonstrated to be a potent negative regulator of T-cell immune function [145]. These findings offer a plausible link between GPR174 polymorphisms and autoimmunity. Polymorphisms in GPR174 have been found to be associated with Graves’ disease [146] [147]. A serine to proline non-synonymous variant in GPR174 has been associated with AAD [89]. The localisation of GPR174 on chromosome X and its role in AAD autoimmunity are particularly interesting given the gender bias observed in this disorder, although it is unlikely that a single gene is responsible for the higher susceptibility of females to develop AAD (Fig. 4.3).
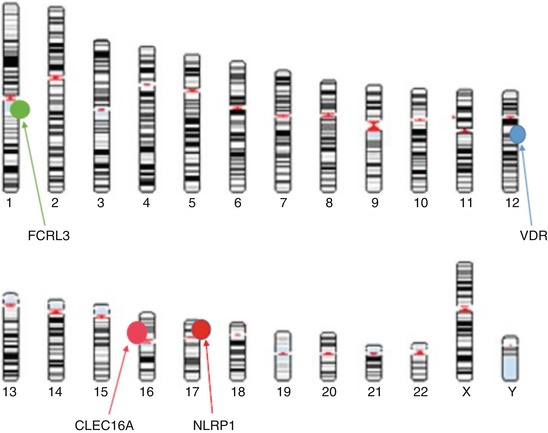
Fig. 4.3
Schematic representation of genes found to be associated with autoimmune Addison’s disease which are implicated in proliferation and activation of B cells and antigen-presenting cells
Genes Implicated in B Lymphocyte and Antigen-Presenting Cell Proliferation and Activation
Vitamin D has been recognised for its effects on the immune system. On a molecular level, the active form of vitamin D, 1,25-dihydroxyvitamin D, leads to reduced expression of HLA class II molecules on endocrine cells and inhibits T-cell proliferation [148, 149]. 1,25-Dihydroxyvitamin D is also implicated in innate immunity by inhibiting differentiation of dendritic cells which are potent antigen-presenting cells [150]. Polymorphisms in the vitamin D receptor (VDR) gene, located on chromosome 12q12-14, have been studied in a number of autoimmune conditions including type 1 diabetes and autoimmune thyroid disease with conflicting results [151–154]. Although specific genotypes in VDR have been associated with AAD risk in a relatively small German cohort, no such association was found for an individual VDR allele [77]. Additional studies are required to confirm VDR as a susceptibility locus for AAD. In contrast to this, a promoter polymorphism in the CYP27B1 gene (−1260C > A) has been shown to be associated with AAD in two independent cohorts [74, 75]. More recently, an intronic SNP, rs4646536, in CYP27B1, was associated with AAD in a large meta-analysis of several European cohorts [76]. CYP27B1 hydro xylase catalyses the conversion of 25-hydroxyvitamin D to its active form, 1,25-dihydroxyvitamin D. A promoter polymorphism in CYP27B1 might affect enzyme transcription and thus the rate of final hydroxylation of 1,25-dihydroxyvitamin D.
The Fc receptor-like 3 gene (FCRL3) located on chromosome 1q21 encodes an orphan cell surface receptor belonging to the immunoglobulin receptor superfamily, expressed predominantly on B lymphocytes. A polymorphism in the FCRL3 promoter region (FCRL3_3*C) has been implicated in susceptibility to rheumatoid arthritis, autoimmune thyroid disease and systemic lupus erythematosus in Asian cohorts [155]. Contrary to the findings in other autoimmune conditions, a study of a UK AAD cohort found that the FCRL3_3*C variant confers protection from the disease [78]. The allele most associated with disease risk in this cohort was found to be FCRL3_3*T (OR = 1.61). Based on functional studies of this locus, FCRL3_3*T is predicted to result in lower promoter activity. Such contradictory findings for one haplotype conferring both protection and disease susceptibility in different populations illustrate the complexity of the genetic underpinning of polygenic autoimmune diseases including AAD.
NLRP1 (nuclear localisation leucine-rich-repeat protein 1 ) is a regulator of the innate immune response. NLRP1 belongs to the NOD-like receptor family and participates in recognising microbial products, such as lipopolysaccharide, and assembly of inflammasomes, cytoplasmic protein complexes mediating pro-inflammatory responses via cytokine activation [156]. Polymorphisms of the NLRP gene have been reported to confer risk for a number of autoimmune conditions including vitiligo [157], type 1 diabetes [86], coeliac disease [158] and rheumatoid arthritis [159]. A coding variant of NLRP1 (Leu155His) has been associated with AAD in two European cohorts [86, 87]. Surprisingly, different alleles were found to confer risk of AAD in Polish (minor allele A) and Norwegian (major allele T) populations.
The CLEC16A (C-type lectin domain family 16) gene encodes a protein of unknown function but which is almost exclusively expressed in immune cells such as dendritic cells, B lymphocytes and natural killer cells. This makes it a plausible susceptibility gene for autoimmunity. A polymorphism in the CLEC16A gene (intronic SNP rs12917716) was found to be associated with AAD in a Norwegian cohort with an OR of 0.71. Comparable effects of CLEC16A SNPs were previously demonstrated in cohorts of subjects with type 1 diabetes (OR 0.65 to 0.83) [160, 161].
Processes Affecting Gene Expression
Gene expression can be altered by both common copy number variation (CNV) and epigenetic modification such as DNA methylation. As a result, different phenotypes can develop despite similar genetic profiles.
CNV in the human genome has been recently identified as a source of genetic diversity and has been shown to influence disease susceptibility [162]. Recently, CNVs in two genes, UGT2B28 and ADAM3A, have been found to be associated with AAD [163]. However, the mechanism by which this association confers susceptibility to the disease remains unknown.
Abnormal DNA methylation is commonly observed in autoimmune disorders. It has been suggested that hypermethylation (addition of methyl groups to oligonucleotides by DNA methyltransferases) of promoter regions silences genes, whereas intronic hypermethylation is involved in gene activation. DNA methylation has been shown to be one of the mechanisms involved in transcriptional control of genes such as FOXP3, Interferon Gamma and AIRE which in turn influence T-cell differentiation and function. A recent study identified multiple hypomethylated gene promoter regions in DNA isolated from CD4 T cells from AAD subjects [164]. A multitude of differently methylated regions have been localised in genes implicated in immune modulation and autoimmunity suggesting that this epigenetic modification plays a role in the immunopathogenesis of this disease. This is likely to be an area of research going forward.
Related posts:
 Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Adrenal Zonation and Development
Adrenal Zonation and Development
 Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Genetics and Pathophysiology of Congenital Adrenal Hyperplasia
Genetics and Pathophysiology of Congenital Adrenal Hyperplasia
 Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


