Fig. 8.1
MRI of supratentorial AT/RT in a 19-month-old boy. (a) Axial T1 with contrast. (b) Axial T2 FLAIR sequence
Some authors have suggested that the presence of wavy, band-like enhancing zone surrounding an area of necrosis may be an imaging marker of AT/RT (Warmuth-Metz et al. 2008; Arslanoglu et al. 2004). Au Yong et al. reviewed 61 pediatric supratentorial tumors and found that the presence of a thick, wavy enhancing wall around a central area of necrosis had a negative predictive value of 95% and a positive predictive value of 63% for AT/RT (Au Yong et al. 2013).
Imaging features of ETMR are less defined, particularly given the histologic variety of these molecularly unified tumors. One recent work reviewed cases from the German HIT studies and compared imaging characteristics of 22 cases each of ependymoblastoma, ependymoma, and CNS-PNET NOS (Nowak et al. 2015). Compared to ependymoma, ependymoblastoma occurred in younger patients, with hypointense instead of isointense T1-weighted imaging with more clearly defined tumor borders and more homogenous enhancement. Large cysts greater than 1 cm diameter were more frequent in ependymoma. Compared to CNS-PNET NOS tumors, ependymoblastoma occurred in younger patients with larger tumors. All of the ependymoblastoma cases, 89% of the CNS-PNET NOS cases, and 80% of the ependymoma cases showed restricted diffusion, suggesting that diffusion-weighted imaging may be useful to discriminate between these high-cellularity tumors and lower-cellularity tumors such as low-grade gliomas.
8.5 Workup
Because patients often present with the rapid onset of symptoms, initial evaluation usually prompts an urgent CT head that identifies a hyperdense mass, with or without additional metastatic sites. Acute management may include steroids to lessen edema and surgical placement of a shunt to address increased intracranial pressure due to obstructive hydrocephalus.
Surgical resection is both diagnostic and therapeutic, as it allows for pathologic diagnosis and also alleviates the mass effect of the tumor on the surrounding tissue. Depending on the clinical scenario, the initial diagnosis of AT/RT may be made after biopsy only or a maximal safe resection of the primary mass.
Histologically, AT/RT is characterized by the presence of rhabdoid cells and development along divergent pathways including neuroectodermal, mesenchymal, and epithelial lines. The defining diagnostic marker of AT/RT is loss of INI1 nuclear staining, which distinguishes AT/RT from other CNS tumors (Dunham 2010).
Correct diagnosis is critical for accurate therapy. Retrospective review of a Children’s Cancer Group study of children less than 3 years old with brain tumors revealed that 10–15% had AT/RT on further pathologic study (Packer et al. 2002). These patients were enrolled prior to mandatory immunohistochemical analysis of tumor samples. In another series, only 12 of 22 patients were correctly diagnosed as having AT/RT based on their initial pathology review; ten patients were initially given other histologic diagnoses and were treated according to protocols for other tumor types (Slavc et al. 2014). Notably, three of those incorrectly diagnosed cases were initially diagnosed as ependymoblastomas.
One-third of AT/RT patients have germline abnormalities of SMARCB1 that can cause rhabdoid tumors outside of the CNS, often in the kidney; these tumors are called malignant rhabdoid tumors (Eaton et al. 2011). The occurrence of two or more rhabdoid tumors in separate sites, such as the brain and kidney, should prompt referral for genetic testing (Eaton et al. 2011). Genetic abnormalities in SMARCB1 are not specific for rhabdoid tumors; they can also be found in choroid plexus carcinoma, sarcomas, and primitive neuroectodermal tumors (Eaton et al. 2011). A family history of schwannomatosis should also trigger genetic evaluation due to its association with germline SMARCB1 alterations (Boyd et al. 2008; Hulsebos et al. 2007; Swensen et al. 2009). These genetic alterations can also give rise to a variety of phenotypic findings, including developmental delay and heart defects (Jackson et al. 2007). Therefore, a detailed family history and clinical evaluation at the time of initial diagnosis of AT/RT are critical to identify patients and families that may be affected by these genetic abnormalities.
Combined LIN28A immunohistochemistry and FISH analysis of the 19q13.42 locus for amplification of the C19MC miRNA cluster are recommended as diagnostic markers for ETMR (Korshunov et al. 2014). LIN28 expression also occurs in gliomas and AT/RT (Deisch et al. 2011; Mao et al. 2013).
After initial diagnosis, complete evaluation includes an MRI of the total spine with and without contrast as well as cerebrospinal fluid (CSF) analysis to determine if disease has disseminated outside of the primary site. Both AT/RT and ETMR have a propensity to spread within the neuroaxis; leptomeningeal spread of an ETANTR is shown in Fig. 8.2. The MRI spine should be obtained preoperatively, within 48 h postoperatively, or at least 2 weeks postoperatively. CSF should be obtained from a lumbar puncture performed at least 2 weeks postoperatively. Based on these results, the extent of disease spread is usually described using a modified Chang staging. Briefly, M0 is no disease outside of the primary site, M1 is microscopic spread to the CSF only, M2 is gross tumor deposits in metastatic sites within the brain, and M3 is gross tumor deposits in the spine (Chang et al. 1969).
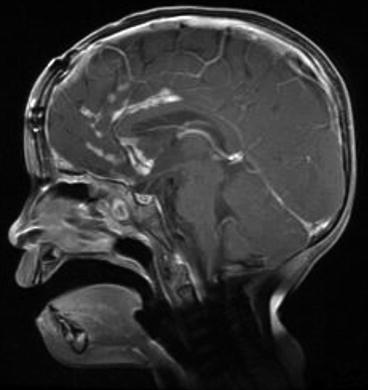
Fig. 8.2
Sagittal T1 with contrast imaging showing leptomeningeal spread of ETANTR in a 3-year-old girl
8.6 Treatment
8.6.1 Surgery
Resection of AT/RT is diagnostic and therapeutic, and the extent of resection correlates with long-term survival outcomes. On the prospective Dana-Farber trial, the 2-year overall survival (OS) for patients undergoing a gross total resection (GTR) was 91%. The median OS for patients with less than a GTR was 18 months (Chi et al. 2009). Similarly, the AT/RT registry found that of 20 patients undergoing GTR, median survival was 20 months compared to 15.25 months for the 22 patients who received partial resection or biopsy alone (Hilden et al. 2004). Based on these data, second-look surgery was encouraged on the COG ACNS0333 protocol for AT/RT patients with tumors that became resectable after induction chemotherapy. Results from that trial are expected to inform future decisions regarding the role of surgery. In the interim, maximal safe resection is the standard of care for AT/RT.
Small case series suggest that extent of resection also correlates with outcomes in ETMR. In a review of 11 cases of ependymoblastoma treated on the prospective HIT trials, the three patients in continuous complete remission all underwent GTR (Gerber et al. 2011). Similarly, in a review of eight patients with medulloepitheliomas, the two long-term survivors both had GTR, with one of those patients undergoing repeat GTR at the time of tumor recurrence (Molloy et al. 1996). Based on these small numbers, maximal safe resection is recommended for ETMR, consistent with treatment of other embryonal tumors.
8.6.2 Chemotherapy
Early approaches to ATRT were based on the third Intergroup Rhabdomyosarcoma Study (IRS-III) protocol for parameningeal rhabdomyosarcoma, Regimen 36. Using this multidisciplinary approach, Weinblatt and Kochen reported on one of the first AT/RT patients with a long-term remission (Weinblatt and Kochen 1992). Similarly, Olsen et al. described three patients treated similarly who also had prolonged survival (Olson et al. 1995). Investigators from the Dana-Farber Cancer Institute (DFCI) then described four patients, two with newly diagnosed disease and two with relapsed disease, treated according to a modified IRS-III protocol that were without evidence of disease at a median of 3 years 1 month after treatment (Zimmerman et al. 2005).
These encouraging results led to the first prospective study in AT/RT, a phase II study led by the DFCI (Chi et al. 2009). After maximal safe resection, patients received pre-irradiation induction chemotherapy, concurrent chemoradiation, post-irradiation induction chemotherapy, maintenance therapy, and continuation therapy. The IRS-III chemotherapy backbone included vincristine, dactinomycin, cyclophosphamide, cisplatin, doxorubicin, and imidazole carboximide (DTIC). On the DFCI protocol, temozolomide was used instead of DTIC because of its ability to penetrate the CNS. The DFCI protocol also included intrathecal chemotherapy with methotrexate, cytarabine, and hydrocortisone. Evaluation prior to radiation therapy yielded a chemotherapy response rate of 58% in 12 evaluable patients. Overall, this aggressive 51-week regimen yielded encouraging results, with a 2-year progression-free survival (PFS) of 53% and OS of 70%.
For children less than 3 years of age, several efforts have been made to avoid or delay radiotherapy. The POG 9233/34 study included 36 patients with AT/RT that were randomized to receive standard or dose-intensified cyclophosphamide and vincristine (Strother et al. 2014). These patients all died of disease, with a median survival of 6.7 months (Ginn and Gajjar 2012). On the CCG-9921 study, children less than 36 months old were treated with vincristine, cisplatin, cyclophosphamide, or etoposide vs. vincristine, carboplatin, ifosfamide, and etoposide. Patients with residual tumor or metastatic disease received delayed radiation; patients without residual tumor did not receive radiation until progression. Of 28 patients with rhabdoid tumors, the 5-year event-free survival was 14% (Geyer et al. 2005). Notably, on both the POG and CCG studies, the diagnosis of AT/RT was made based on the histologic appearance of the tumor, not on molecular testing.
Given these results with conventional chemotherapy, high-dose chemotherapy (HDC) with stem cell rescue (SCR) has been pursued to intensify therapy while still avoiding or delaying radiation. On the Head Start I, postoperative patients underwent five cycles of induction with cisplatin, vincristine, cyclophosphamide, and etoposide. This was followed by consolidation with carboplatin, thiotepa, and etoposide with SCR. Head Start II was similarly designed with the addition of high-dose methotrexate to each cycle of induction chemotherapy. (Gardner et al. 2008). All six patients on Head Start I died of disease. Three of seven patients treated on Head Start II were alive without disease, yielding a 3-year EFS of 43%. These results support a role for methotrexate in the treatment of AT/RT.
On Head Start III, cisplatin and high-dose methotrexate alternated with temozolomide for five cycles of induction chemotherapy that also included cyclophosphamide, vincristine, and etoposide. This was followed by second-look surgery and consolidation with HDC with SCR. Patients 6–10 years old or with residual tumor prior to consolidation received radiation after consolidation. Of 19 patients enrolled, five died from toxicity during induction. Only four patients completed the five planned cycles of induction therapy, and three of those patients completed HDC with SCR. One of those patients had a continuous complete response, and two had partial responses. The 3-year EFS and OS were 21% and 26% (Zaky et al. 2014).
Based on the results described above, the COG study ACNS0333 includes two cycles of intense induction chemotherapy, similar to the POG9923 and CCG-9921 approaches, with systemic methotrexate as in Head Start II. Consolidation includes three cycles of HDC with carboplatin and thiotepa with SCR, based on results from CCG 99703. The COG study is currently closed to accrual and results are anticipated to direct development of the next generation of systemic therapies for AT/RT.
The optimal chemotherapy approach for ETMR remains unknown. From an analysis of 36 patients with PNET of the CNS with C19MC amplification and LIN28 positivity, Spence et al. found that patients receiving chemotherapy with or without radiation had a median survival of 13 months compared to 0.06 months for patients receiving no therapy (p = 0.004) (Spence et al. 2014). In published case report in the literature, patients have generally been treated according to CNS-PNET protocols or protocols designed for infants with malignant brain tumors such as CCG 99703 (Lafay-Cousin et al. 2014). From a review of 11 patients with ependymoblastoma treated on the HIT protocols, one patient survived after HDC without radiation, one patient had a response after conventional chemotherapy, and four patients had early relapses during induction chemotherapy prior to RT. Based on these observations, the authors suggest that at least for patients with residual disease after surgery, induction chemotherapy should be relatively short (Gerber et al. 2011). There is also a case report of transient maturation of ETMR after chemotherapy delivered according to CCG 99703 (Lafay-Cousin et al. 2014). Although the patient subsequently developed recurrent disease, her overall survival from diagnosis was 21 months, which is encouraging.
8.6.3 Radiation Therapy
Because of the young age of most AT/RT patients, the use of radiation for this tumor has been controversial (Squire et al. 2007). Early efforts focused on approaches that delayed radiotherapy, based on results from medulloblastoma that chemotherapy can delay radiation until after 3 years of age (Duffner et al. 1993). One of the earliest series in AT/RT came from a registry that followed 42 patients. Of 13 patients who received radiation, eight were alive without evidence of disease at least 19 months from diagnosis (Hilden et al. 2004). In a review of the St. Jude’s experience, ten children treated with chemotherapy and radiation had a 2-year OS estimate of 90% vs. 12% for 21 children treated without radiation (Tekautz et al. 2005).
A recent review of 12 publications describing 332 children and adolescents with AT/RT found that initial radiotherapy was associated with improved RFS and OS (Schrey et al. 2016). Of 238 patients with radiation data, 118 had received either focal, cranial, or craniospinal radiation. The median RFS of patients who received radiation was 12.1 months, compared to 4 months for patients not receiving radiation (p < 0.001). Similarly, the median OS of patients receiving radiation was 23 months vs. 10 months for patients treated without initial radiation (p < 0.001). The association of upfront radiation with significantly improved RFS and OS was upheld on multivariate analyses including intrathecal chemotherapy, extent of surgery, and HDC with SCR. Based on emerging evidence supporting a role for radiation in AT/RT, the use of radiation has increased from 22.3% of cases diagnosed between 1973 and 2004 to 38.4% of cases since 2005 (Lau et al. 2015).
The optimal use of radiation in ETMR is unknown. In a literature review by Alexiou et al., 17 of 72 patients received radiation and had a better median OS compared to patients who did not receive radiation, 16 months vs. 11 months (p = 0.029) (Alexiou et al. 2013). Mozes et al. reviewed cases of seven children with recurrent ETANTR and found that the four patients who received radiation had better mean OS than the three who did not, 31.5 months vs. 24.0 months (Mozes et al. 2016). Based on these results, a role for radiation in ETMR is emerging, but clearly more data are needed.
8.6.4 Radiation Fields and Doses
Optimal radiation treatment fields for ATRT remain unknown. In the initial DFCI report, patients with M0 disease at presentation received local radiation, and patients older than 3 years with M+ disease received CSI (Chi et al. 2009). CSI was delivered to 36 Gy and local fields were treated to 54 Gy. All patients received intrathecal chemotherapy although it is unclear if intrathecal chemotherapy can substitute for CSI.
On the COG study ACNS0333, patients with M0 disease at least 6 months old with an infratentorial tumor or at least 12 months old with a supratentorial tumor received focal RT after two cycles of induction chemotherapy. Younger M0 patients received focal RT after completing induction and consolidation chemotherapy. Patients with M+ disease at enrollment were encouraged but not mandated to receive age-adjusted CSI after induction and consolidation chemotherapy. For patients with M0 disease younger than 36 months, local fields were treated to 50.4 Gy, and for patients older than 36 months, local fields were treated to 54 Gy. Patients with M+ disease younger than 36 months were treated with CSI to 23.4 Gy and patients older than 36 months were treated with CSI to 36 Gy. All treatment was delivered at 1.8 Gy/fraction, one fraction per day.
Patients at least 3 years old were eligible for SJMB03, on which all ATRT patients received CSI after surgery. Patients with M0 disease and less than 1.5 cm3 residual tumor were classified as average risk and treated with CSI to 23.4 Gy followed by a boost to the tumor bed to 55.8 Gy. Patients with M+ disease or more than 1.5 cm3 residual tumor were classified as high risk and received 36–39.6 Gy CSI. After a 6-week rest period, all patients received four cycles HDC with peripheral blood stem cell support.
Results from ACNS0333 and SJMB03 are awaited to better define optimal radiation fields for ATRT, especially for M0 patients. In our retrospective review of our experience treating 31 patients with ATRT with proton therapy, one patient failed on the margin of the radiation field and seven patients failed in the neuroaxis but outside of the high dose volume (McGovern et al. 2014). These results demonstrate the efficacy of radiation in AT/RT, but especially in very young patients the risk of radiation exposure must be balanced against the long-term effects of radiation. Proton therapy may alter this toxicity equation by decreasing radiation dose to normal tissues. Results from MD Anderson (McGovern et al. 2014), the Paul Scherrer Institute (Weber et al. 2015) and Massachusetts General Hospital (De Amorim et al. 2013) show that proton therapy for AT/RT in young patients is well tolerated with acceptable short-term side effects; longer follow-up is clearly needed in this population.
Radiation fields and doses for ETMR are not defined but are typically based on those for PNET or AT/RT. In our institutional review of seven patients with ETMR treated with proton therapy, the median age at radiation start was 42 months (range, 17–58 months). Two patients received local fields only to 50.4 or 54 Gy(RBE) and five received CSI to 36 Gy(RBE) followed by a tumor bed boost to 45–55.8 Gy(RBE).
8.7 Outcomes
After completion of therapy, surveillance of patients with AT/RT or ETMR is similar to that for patients with PNET. Follow-up should include MRI of the brain and spine every 3 months for the first 1–2 years after therapy, then every 6–12 months thereafter. Patients are at risk for early and late recurrence in the CNS. Progression prior to initiation of radiation is well documented and portends a poor prognosis (Chi et al. 2009; McGovern et al. 2014). In addition to the risk of recurrence, patients are at risk for the sequelae of treatment and should be followed for specific toxicities including neurocognitive, endocrine, auditory, and visual deficits. Guidelines for long-term follow-up for pediatric survivors of CNS tumors have been published by the COG (Group CsO 2014).
Historically, survival in AT/RT has been bleak, typically less than 1 year (Burger et al. 1998). In the most recent CBTRUS analysis of SEER data from 2007 to 2011, OS in ATRT was 48.1%, 27.5%, and 26.0% at 1, 5, and 10 years (Ostrom et al. 2015). Radiation may contribute to improved outcomes in more recent studies. On the DFCI 02-0294 study, in which 15 of 20 evaluable patients received radiation, the 2-year PFS and OS were 53% and 70% (Chi et al. 2009). As detailed above, a recent literature review of 332 cases of AT/RT also found that initial radiation was associated with improved RFS and OS (Schrey et al. 2016). Results from ACNS0333 and SJMB03 are awaited for additional prospective survival data.
Patients with familial AT/RT tend to have worse outcomes than those with sporadic disease. In a retrospective review of 20 patients with AT/RT, children with familial AT/RT were diagnosed at a median age of 4.8 months and had a median OS of 4.5 months. This was markedly worse than patients with sporadic AT/RT, who were diagnosed at a median age of 13 months and had a median OS of 21 months (Bruggers et al. 2011). The younger age of children with familial disease limits therapeutic options and likely contributes to their bleak outcomes.
Outcomes for ETMR tend to be worse than for classic supratentorial PNET. Of 11 children with ependymoblastoma treated on the prospective German HIT trials, 5-year PFS was 36.4% and 5-year OS was 30.3% (Gerber et al. 2011). The four survivors in this group had either craniospinal radiation and/or HDC-SCR, suggesting that multi-modality therapy may contribute to better outcomes for these patients. Preliminary data from a literature review from our institution identified 178 reported cases of ETMR with a median OS of 10 months from diagnosis. Eighteen of these patients were long-term survivors, with a median survival of 77.2 months from diagnosis; 17 of the 18 long-term survivors received radiation.
8.8 Toxicities
The aggressive multi-modality therapy used to treat patients with AT/RT and ETMR can cause long-term effects, affecting a variety of body systems. Because of the very young age of most patients, neurocognitive outcomes are a primary concern. Even in the absence of radiation therapy, long-term survivors of AT/RT can have neurocognitive deficits, as demonstrated in a cohort of 11 patients in the Canadian Registry (Lafay-Cousin et al. 2015). This observation was upheld in a larger study of 224 patients from St. Jude’s, which found that long-term survivors of pediatric CNS tumors had neurocognitive impairment compared to normative national data, even in patients that received no radiation. The degree of impairment varied with the volume of radiation exposure. For instance, in a measure of long-term memory, 11% of survivors with no cranial radiation showed impairment compared to 25% of patients treated with focal cranial RT and 36% exposed to CSI (Brinkman et al. 2016). Long-term survivors who have received radiation are also at risk for pulmonary, endocrine, cardiovascular, and gastrointestinal dysfunction, especially after CSI (Huang et al. 2014; Saha et al. 2014).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


