Fig. 16.1
Three-dimensional structure of VDAC1 and its channel activity. (A), VDAC1 monomer and dimer structures: (a) Side view of the crystal structure of VDAC1 (PDB code: 3EMN). The β-barrel is formed by 19 β-strands and the N-terminal helix is folded into the pore interior. Both the N- and C-termini (N and C, respectively) are located on the same side of the membrane. The N-terminus is colored red. (b) Top view of VDAC1 with the N-terminal helix nested inside the VDAC1 pore. (c) A proposed model for the conformation of VDAC1 with its N-terminal on the outside of the VDAC1 pore. The LP4 and (ΔN1-14)N-Terminal domains used as anti-cancer peptides are depicted in blue (see Sect. 1.7). (d) A proposed dimer of VDAC1. Figures were prepared using PyMOL software. (B, C) Bilayer-reconstituted VDAC1 single and multi-channel activity was assayed as described previously (Arbel et al. 2012). (B) shows a typical current recording through VDAC1 (1 M NaCl) in response to voltage steps from 0 to 10 mV showing a constant conductance, and from 0 to 60 mV, with the current first increased (m) due to a greater driving force then decreased due to a conformational change to a low-conducting state (s). (C) The average steady-state conductance of VDAC1 is presented as a function of voltage. Relative conductance was determined as the ratio of conductance at a given voltage (G) and the maximal conductance (G0) at 10 mV, showing the bell-shape voltage-dependence characteristic of VDAC1. (D) Mitochondria-purified VDAC1 that was used in the PLB experiments
16.1.2.2 Channel Properties
The channel properties of purified VDAC1 have been examined following reconstitution of the purified protein into a planar lipid bilayer (PLB), using various procedures and detergents (Shoshan-Barmatz et al. 2010a). Bilayer-reconstituted VDAC1 assumes multiple voltage-dependent conformational states (Fig. 16.1B) displaying different selectivities and permeabilities. VDAC1 shows symmetrical bell-shaped voltage-dependent conductance (Fig. 16.1C) with the highest conductance (4 nS at 1 M KCl) occurring at low potentials of −20 to +20 mV (Colombini 2012). At low potentials, and when in the fully open state, VDAC1 selectively conducts small ions (e.g. Cl−, K+, Na+), yet shows a preference for anions, such as phosphate, chloride, adenine nucleotides, glutamate, and other anionic metabolites, and large cations, such as acetylcholine, dopamine and Tris (Shoshan-Barmatz et al. 2010a). At higher positive or negative potentials (>30–60 mV), channel conductance is reduced and the selectivity shifts to small cations. In this scenario, the channel becomes virtually impermeable to ATP and ADP (Colombini 2012; Shoshan-Barmatz et al. 2010a).
As a voltage-gated channel, it is believed that VDAC1 channels rely on two separate gating processes, one at positive trans-membrane potentials and the other at negative potentials (Colombini 2012; Shoshan-Barmatz et al. 2010a). The N-terminal α-helical segment of the channel has been proposed to act as the voltage sensor, gating the pore via conformational changes and/or movements (Sect. 1.2.1). Clearly, additional studies are required before the molecular nature of the VDAC1 gating mechanism can be resolved.
16.1.3 VDAC Functions Are Important for Cancer Cells
VDAC is a 31 kDa pore-forming protein found in the outer mitochondrial membrane (OMM) of all eukaryotes (Colombini 2012; Shoshan-Barmatz et al. 2010a). Three eukaryotic VDAC isoforms, encoded by three separate genes sharing 65–70 % of sequence homology, VDAC1, VDAC2 and VDAC3, have been identified (for review, see (Shoshan-Barmatz et al. 2010a)). All VDAC isoforms can be found in most tissues, albeit at different levels of expression, with the most abundant and studied being VDAC1 (Shoshan-Barmatz et al. 2010a). The specific role of each isoform remains unclear, although evidence indicates that the three isoforms may serve different physiologic functions (Shoshan-Barmatz et al. 2010a). The focus of this review is on VDAC1.
16.1.3.1 VDAC1 Transport Activity Can Control the Fate of the Cell
At the OMM, VDAC1 functions as gatekeeper for the entry and exit of mitochondrial metabolites, assuming a crucial position in the cell, serving as the main interface between mitochondrial and cellular metabolisms. VDAC1 thus mediates the fluxes of ions, nucleotides and other metabolites up to ~5,000 Da across the OMM (Shoshan-Barmatz et al. 2010a) (Fig. 16.2A). Moreover, its location at the boundary between the mitochondria and the cytosol enables VDAC1 to interact with proteins that mediate and regulate the integration of mitochondrial functions with other cellular activities (Shoshan-Barmatz and Golan 2010; Shoshan-Barmatz et al. 2006, 2010a, 2013; Shoshan-Barmatz and Mizrachi 2012).
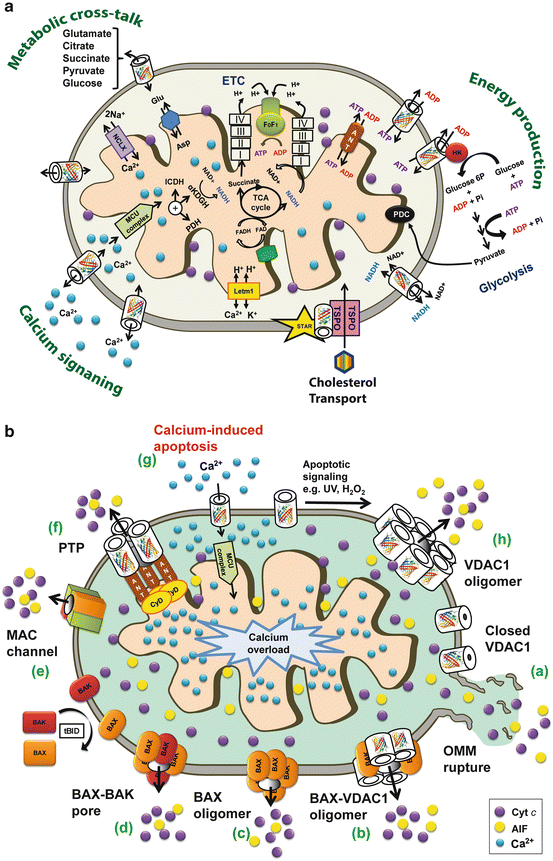
Fig. 16.2
Schematic representation of VDAC1 as a multi-functional channel essential for cancer cell survival and regulator of cell death. (A) VDAC1 functions in cell life – The various functions of VDAC1 include control of the metabolic cross-talk between the mitochondria and the rest of the cell, cellular energy production by transporting ATP/ADP and NADH between the Fig. 16.2 (contiuned) inter-membrane space (IMS) and the cytosol, binding HK, Ca2+ signaling by transporting Ca2+, and cholesterol transport. Also presented are the Ca2+ influx and efflux transport systems of the OMM and IMM, as well as Ca2+-mediated regulation of the tricarboxylic acid (TCA) cycle via activation of pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH) and α-ketoglutarate dehydrogenase (α KGDH). The electron transport chain (ETC) and the ATP synthase (FoF1) are also presented. VDAC1 in the OMM transports Ca2+ to the IMS. The uptake of Ca2+ into the matrix via the IMM is mediated by the mitochondrial Ca2+ uniporter (MCU), regulated by a calcium-sensing accessory subunit (MICU1). Ca2+ efflux is mediated by NCLX, a Na+/Ca2+ exchanger. High levels of matrix Ca2+ accumulation trigger the opening of the permeability transition pore (PTP), a fast Ca2+ release channel. (B) VDAC1 function in cell death – Different models for the release of apoptogenic proteins, such as Cyto c (purple circles) and AIF (yellow circles) from the IMS to the cytosol, leading to apoptosis. These models include: (a) VDAC1 closure and OMM rupture serving as the Cyto c release pathway; (b) A Bax- and VDAC1-based hetro-oligomer mediating Cyto c release; (c) Bax activation followed by its oligomerization resulting in OMM permeabilization; (d) A pore formed by oligomerized forms of Bax and Bak; (e) MAC forms during the early apoptosis stage as the release pathway; (f) A PTP composed of VDAC1 at the OMM, ANT at the IMM and CypD in the matrix providing the apoptogenic protein release pathway; (g) Mitochondrial Ca2+ overload induces apoptosis – Ca2+ transport across the OMM, as mediated by VDAC1, and then across the IMM, as mediated by the MCU, leads to Ca2+ overload in the matrix. This is turn causes dissipation of the membrane potential, mitochondria swelling, PTP opening, Cyto c release and the triggering of apoptotic cell death; (h) A VDAC1 homo-oligomer forming the apoptotic proteins-conducting channel (see Sect. 1.5)
VDAC1 Transport Activity Controls Energy and Metabolites
In the open state, VDAC1 allows free shuttling of ATP and ADP as well as NAD+/NADH. Mitochondria-generated ATP is transported to the cytosol for exchange with ADP, which is utilized in oxidative phosphorylation to generate ATP. As such, VDAC1 controls the electron transport chain, helping to generate energy and support survival (Shoshan-Barmatz et al. 2010a). VDAC1 closure limits the normal flow of metabolites in and out of mitochondria and thus impairs cellular metabolic and energy homeostasis (Vander Heiden et al. 2000). Indeed, silencing VDAC1 expression resulted in reduced metabolite exchange between mitochondria and the cytosol, showing VDAC1 to be essential for energy production and cell growth (Abu-Hamad et al. 2006; Arif et al. 2014).
VDAC1 function in energy metabolism is also reflected in its interaction with hexokinase (HK) and creatine kinase (CK) to convert newly generated ATP into high-energy storage forms, such as glucose-6-phosphate and creatine phosphate, respectively (Shoshan-Barmatz et al. 2010a). In keeping with its two-way trafficking role, VDAC1 also enables the exit of newly formed hemes, as well as the entrance of substrates of the electron transport chain, including pyruvate, malate, succinate and NADH (Shoshan-Barmatz et al. 2010a).
VDAC1 in Cholesterol Transport
In general, cancer cells have been shown to exhibit two- to ten-fold more mitochondrial cholesterol (mainly in the OMM) than found in liver mitochondria, thus altering the fluidity of the cancer cell membrane (Yu et al. 2005). Cholesterol is transported across the OMM (Fig. 16.2A) (Rone et al. 2009), with VDAC1 being considered to be a necessary component of a multi-protein complex, the transduceosome, thought to also contain the high-affinity cholesterol-binding protein translocator protein (TSPO) and the steroidogenic acute regulatory protein (STAR) (Campbell and Chan 2008). TSPO interacts with VDAC1, helping to anchor the multi-protein complex to the OMM and assists with the binding and import of STAR (Miller 2013), hence serving as the acute regulator of steroidogenesis. In addition, high cholesterol affects HK binding to VDAC1, and accordingly, the metabolic function of VDAC1 (Campbell and Chan 2008). Thus, VDAC1 affects both cholesterol synthesis and transport, and is subject to cholesterol-mediated regulation.
VDAC1 as a ROS Transporter
Reactive oxygen species (ROS) are well-known to play a part in proliferation and cell death, with cellular levels of ROS being linked to anti-tumor immunity, the oxidative tumor micro-environment, the proliferation and death of cancer cells (Manda and Neagu 2009). Hypoxia, a characteristic of most solid tumor micro-environments, causes a progressive elevation in mitochondrial ROS production (chronic ROS) which activates the transcription of genes involved in cellular hypoxic adaptation (Hamanaka and Chandel 2009).
Mitochondria is a major source of ROS in the cell that attacks DNA, lipids and proteins, thereby affecting cell survival (Handy and Loscalzo 2012). ROS release to the cytosol is mediated by VDAC1, with such transport being regulated by VDAC1-bound HK-I and HK-II, serving to reduce intracellular levels of ROS (Fig. 16.2B) (da-Silva et al. 2004). Closure of VDAC1 causes oxidative stress and accelerates Ca2+-induced opening of the mitochondrial permeability transition pore (PTP) (Tikunov et al. 2010). As mitochondrial dysfunction is one of the important features of ROS-mediated cell death, the ROS release function of VDAC1 is an important activity protecting against mitochondrial damage.
In summary, VDAC1 appears to be a convergence point for a variety of cell survival signals regulating the metabolic and energetic functions of mitochondria by its transport activity, as well as by its association with various ligands and proteins. As such, VDAC1 can control the fate of cancer cells (Abu-Hamad et al. 2009; Abu-Hamad et al. 2006).
16.1.4 Cancer Cell Metabolism and Targeting VDAC1 by siRNA to Inhibit Cell and Tumor Growth
In recent years, a substantial body of evidence has accumulated indicating a correlation between alterations in cell metabolism and cancer formation (Gatenby and Gillies 2004; Hanahan and Weinberg 2011; Koppenol et al. 2011; Shoshan-Barmatz and Golan 2010). Cancer cells undergo significant metabolic adaptation to fuel cell growth and division (Gatenby and Gillies 2004; Hanahan and Weinberg 2011; Koppenol et al. 2011). Malignant cancer cells typically display high rates of glycolysis even when fully oxygenated and are subject to suppressed mitochondrial respiration, despite the fact that glycolysis is a less energy-efficient pathway, a phenomenon known as the ‘Warburg effect’ (Gatenby and Gillies 2004; Hanahan and Weinberg 2011; Koppenol et al. 2011). The Warburg effect likely provides the vast majority of cancerous cells with a number of benefits in the form of precursors for the biosynthesis of nucleic acids, phospholipids, fatty acids, cholesterol and porphyrins. A second advantage of the Warburg effect is its expected involvement in both tumor protection and invasion. Tumor cells produce lactic acid via glycolysis and transport it out of the cell, leading to increased acidity of the closed micro-environment and the generation of a low pH ‘coat’. This coat is proposed to protect tumors against attack by the immune system while inducing negative effects on normal surrounding cells, aiding in preparing the surrounding tissues for invasion. Additionally, the Warburg effect also assures longer tumor survival time if oxygen becomes limiting (Hanahan and Weinberg 2011). Moreover, cancer-associated abnormalities in glucose metabolism enhance cellular resistance to apoptosis, with mitochondria playing a key role in this process (Fulda et al. 2010; Gogvadze et al. 2010; Kroemer and Pouyssegur 2008; Mayevsky 2009). Finally, mitochondria have been found to contribute to cellular re-programming from the catabolic to the anabolic mode (Gogvadze et al. 2010; Kroemer and Pouyssegur 2008; Mayevsky 2009), with such metabolic flexibility and cellular hierarchy being crucial in metastatic cancer (Berridge et al. 2010).
VDAC1 functions are indispensable for proper mitochondria functions and, consequently, for cell activity. Specifically, VDAC1 is crucial for a range of cellular processes, including ATP rationing, Ca2+ homeostasis and apoptosis execution (Shoshan-Barmatz et al. 2006, 2010a, 2013; Shoshan-Barmatz and Golan 2010; Shoshan-Barmatz and Mizrachi 2012 (Fig. 16.2B). These activities are regulated via the interaction of VDAC1 with many proteins central to the regulation of cell survival and cellular death pathways (Shoshan-Barmatz et al. 2006, 2010a, 2013; Shoshan-Barmatz and Golan 2010; Shoshan-Barmatz and Mizrachi 2012). VDAC1 serves as a key binding target for nearly two dozen proteins (see Sect. 1.6), (Hanahan and Weinberg 2011; Shoshan-Barmatz et al. 2006; Shoshan-Barmatz and Mizrachi 2012). These interactions point to VDAC1 as a convergence point for a variety of cell survival and cell death signals, and thus interfering with these interactions could impair cell homeostasis, as desired in case of cancer cells.
16.1.4.1 Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo
The over-expression of VDAC1 in some cancer cells may be related to its multi-functional activities required by the high-energy demands of cancer cells (Shoshan-Barmatz et al. 2010a). As such, down-regulation of VDAC1 expression results in reduced metabolite exchange between mitochondria and the cytosol, leading to mitochondrial dysfunction and arrest of cell growth, demonstrating the essential role of VDAC1 in energy production and cell growth (Abu-Hamad et al. 2006; Koren et al. 2010). Similarly, alterations of mitochondrial function are linked to VDAC1 closure, which limits the normal flow of metabolites in and out of mitochondria (Vander Heiden et al. 2000). Indeed, previous studies from our lab have demonstrated that down-regulation of VDAC1 expression by hVDAC1-shRNA disrupts energy production and cell growth and inhibits tumor development in an animal model (Abu-Hamad et al. 2006; Koren et al. 2010).
Diminished VDAC1 expression by means of shRNA strongly inhibited cell proliferation and cancer cell growth in vitro in cell cultures, and in vivo using animal models (Abu-Hamad et al. 2006; Koren et al. 2010). siRNA at nanomolar concentrations silenced VDAC1 expression in all tested cell lines, including lung cancer A549, prostate cancer PC-3, glioblastoma U87 and hepatocellular carcinoma HepG2 cells, leading to a remarkable decrease in VDAC1 protein levels and inhibition of cancer cell growth (over 90 %), which persisted up to 144 h post-transfection (Fig. 16.3A, B) (Arif et al. 2014). Cells expressing low VDAC1 levels contained low ATP levels, suggesting limited metabolite exchange between mitochondria and cytosol (Abu-Hamad et al. 2006; Koren et al. 2010).
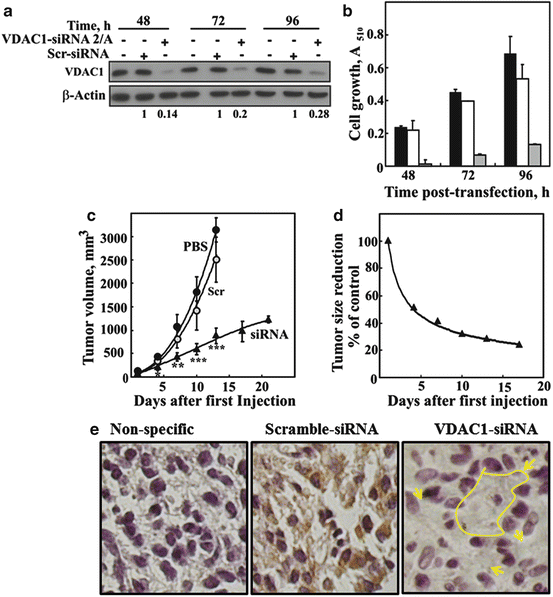
Fig. 16.3
Modified hVDAC1-siRNA-mediated inhibition of cell growth in vitro and in vivo. U87MG cells were transfected with 50 nM siRNA-hVDAC1 or scrambled siRNA and VDAC1 expression levels (relative units, RU) were evaluated by immunoblot at 48, 72 and 96 h post-transfection (a). Similarly, cell growth was assayed using the SRB method (b), with black, grey and white bars representing non-transfected cells and cells transfected with scrambled or hVDAC1-siRNA, respectively (n = 3). In a xenograft mouse model (c, d), U87MG cells were inoculated into male nude mice (2 × 106 cells/mouse). Tumor volumes were monitored (using a digital caliper) and on day 20, the mice were divided into three groups (eight or nine mice per group), with each group containing a similar average tumor volume (120 mm3). The three mice groups were subjected to the following treatments. Xenografts were injected at two points with PBS (●, control), with scrambled siRNA (o) or with VDAC1-siRNA (▲) (10 μl of a 400 nM solution to yield a final concentration of 40–60 nM of each siRNA). Xenograft sizes as a function of time following the start of treatment is presented in (c), while the fold decrease in tumor size of the VDAC1-siRNA-injected xenografts, as compared to PBS-injected mice, is shown in (d). The calculated average tumor volumes are presented as means ± SEM, P < 0.01 (**) or < 0.001 (***). (e) Histological analysis of paraffin sections cut from tumors removed from scrambled- and VDAC1-siRNA-treated mice was carried out by hematoxylin/eosin and immunohistochemical staining with anti-VDAC1 antibodies, recognizing both mouse Fig. 16.3 (contiuned) and human proteins. Representative sections from each group revealed strong staining in scrambled siRNA-injected tumors, with homogenous and strong staining being seen with anti-VDAC1 antibodies. The sections from the hVDAC1-siRNA -injected tumor showed non-homogenous staining, with strong staining representing a tumor containing U87 cancer cells while some non-stained areas most likely represent cells of mouse origin (marked arrows). NS represents staining with only secondary antibodies. Bars represent 5 μm
In vivo experiments using xenograft lung cancer (Arif et al. 2014) and glioblastoma (Fig. 16.3C, D) mouse models showed that chemically modified VDAC1-siRNA not only inhibited tumor growth but also resulted in tumor regression (Arif et al. 2014). Moreover, immunohistochemical staining with anti-VDAC1 antibodies of tumor sections from scrambled- and VDAC1-siRNA-treated mice showed strong staining of untreated tumor sections as expected from cancer cells, while very weak staining was seen in the treated tumor (Fig. 16.3E) The discriminatory effects of siRNA on cancerous and non-cancerous cancer cells (Arif et al. 2014) may result from the high expression levels of VDAC1 in tumors, suggesting VDAC1 to be important for cancer cell development and survival. Thus, siRNA-VDAC1-mediated inhibition of cancer cell growth and tumor development as a result of disabling the abnormal metabolic behavior of cancer cells points to an approach to treat cancer.
16.1.5 Apoptosis, VDAC1 and Cancer
The mitochondria-mediated apoptotic pathway can be triggered by diverse intracellular signals, such as oxidative stress, chemotherapeutic drugs, Ca2+ overload and DNA damage, and is activated when the pro-apoptotic signals overcome anti-apoptotic signals, leading to mitochondria permeabilization and release of inter-membrane (IMS) apoptogenic proteins (e.g. cytochrome c (Cyto c), AIF, Smac/DIABLO) (Shoshan-Barmatz et al. 2010b; Zaid et al. 2005). The released Cyto c, as a constituent of the apoptosome, activates cysteine-aspartic protease caspase-9 that in turn activates the executioner caspases, caspase-3 and -7, leading to cell destruction (Kroemer et al. 2007).
It remains unclear how apoptotic initiators that reside in the IMS cross the OMM and released into the cytosol. Several models have been proposed, such as the formation of the PTP, the assembly of a protein channel composed of Bax, Bak or both, of Bax and VDAC1, or of VDAC1 alone (Fig. 16.2B). Indeed, it is now recognized that VDAC1 acts as a key player in mitochondria-mediated apoptosis (Lemasters and Holmuhamedov 2006; Shoshan-Barmatz et al. 2006, 2010a, 2013; Shoshan-Barmatz and Golan 2010; Shoshan-Barmatz and Mizrachi 2012). VDAC1 participates in apoptosis via the release of mitochondrial pro-apoptotic proteins to the cytosol, and interacts with apoptosis regulatory proteins, such as Bcl-2, Bcl-xL (Arbel et al. 2012; Arbel and Shoshan-Barmatz 2010; Malia and Wagner 2007; Shimizu et al. 2000; Sugiyama et al. 2002; Tsujimoto and Shimizu 2002) and HK (Abu-Hamad et al. 2009; Abu-Hamad et al. 2008; Arzoine et al. 2009; Zaid et al. 2005) (see Sect. 1.6), that are over-expressed in many cancers (Grobholz et al. 2002; Mathupala et al. 2009). The proposed VDAC1 structure mediating Cyto c release corresponds to a VDAC1 oligomer, forming a channel large enough to enable release of Cyto c and leading to apoptosis induction (see Sect. 1.5.1).
16.1.5.1 VDAC1 Oligomerization, Function, Modulation and Apoptosis Induction
Purified and membrane-embedded VDAC1 were shown to assemble into dimers, trimers, tetramers and higher order oligomers, as revealed by chemical cross-linking and fluorescence resonance energy transfer (FRET) analysis (Keinan et al. 2010; Shoshan-Barmatz et al. 2006, 2010a, 2013; Shoshan-Barmatz and Golan 2010; Shoshan-Barmatz and Mizrachi 2012; Zalk et al. 2005). In addition, the NMR-based structure of recombinant hVDAC1 implied that it forms a dimer of monomers arranged in parallel (Bayrhuber et al. 2008) (Fig. 16.1Ad), while analysis of the crystal packing of mVDAC1 revealed strong anti-parallel dimers that further assemble into hexamers (Ujwal et al. 2009).
We have demonstrated that VDAC1 oligomerization is highly increased upon apoptosis induction, as revealed by chemical cross-linking, or as directly monitored in living cells using BRET (bioluminescence resonance energy transfer), and is accompanied by conformational changes in the protein (Keinan et al. 2010; Shoshan-Barmatz et al. 2008). Enhancement of VDAC1 oligomerization was obtained regardless of the cell type or apoptosis inducer used, including STS, curcumin, As2O3, etoposide, cisplatin, selenite, TNF-α, H2O2 or UV (see also Fig. 16.6B), all capable of activating mitochondria-mediated apoptosis, yet acting through different mechanisms (Keinan et al. 2010). Moreover, VDAC1 over-expression resulted in VDAC1 oligomerization and apoptosis in the absence of any apoptotic stimulus (Shoshan-Barmatz et al. 2008; Weisthal et al. 2014). Structural and computational-based approaches, in combination with site-directed mutagenesis, cysteine replacement and chemical cross-linking, identified contact sites between VDAC1 molecules in dimers and higher order oligomers (Geula et al. 2012b). These and other findings (Abu-Hamad et al. 2009; Keinan et al. 2010; Shoshan-Barmatz et al. 2006, 2008b; Zalk et al. 2005) have led to the proposal that VDAC1 oligomers form large protein-conducting channels, offering the pathway for Cyto c release. This proposed mechanism is further supported by the findings that apoptosis inducers up-regulate VDAC1 expression levels, shifting monomeric to oligomeric VDAC1, leading to Cyto c release and subsequently, to apoptosis (see Sect. 1.8.2). The oligomerization of VDAC1 associated with apoptosis induction provides a new approach for developing a new class of drugs, directly targeting VDAC1 to induce its oligomerization.
16.1.6 Cancer Cells Avoid Apoptosis
Many malignant cells arise from the multi-step process of tumorigenesis, involving accumulation of inherited or acquired genetic alterations that protect malignant cells from apoptosis (Hanahan and Weinberg 2011). In fact, highly aggressive treatment-resistant tumors employ multiple pathways to avoid apoptosis (Hanahan and Weinberg 2011). Over-expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL is seen in numerous cancer types, with the expression of these proteins correlating with resistance to chemotherapy-induced apoptosis (Adams and Cory 2007; Llambi and Green 2011). Thus, specific induction of apoptosis in cancer cells is an effective approach for killing cancer cells. However, conventional apoptosis-inducing chemotherapy is limited by a lack of specificity, resistance, and toxicity to normal cells. Therefore, the potential benefits of developing novel, target-specific anti-cancer drugs interfering with anti-apoptotic proteins interacting with VDAC1 are enormous.
16.1.6.1 VDAC1-Interacting Proteins
VDAC1, located in the OMM, serves as an anchor protein for a diverse set of molecules that interact with the mitochondria. VDAC1 displays binding sites for glycerol kinase, HK, glyceraldehyde 3-phosphate dehydrogenase, creatine kinase, C-Raf kinase, ANT (adenine nucleotide translocase), TSPO (translocator protein), tubulin, the dynein light chain (mtHSP70), the ORDIC channel, gelsolin, actin and superoxide dismutase 1 (SOD1), as well as Bcl-2 family members (Hanahan and Weinberg 2011; Shoshan-Barmatz et al. 2006, 2010a, 2013; Shoshan-Barmatz and Mizrachi 2012). Serving as an anchor point for apoptosis-regulating proteins, such as HK and Bcl-2 family members, some of which are also highly expressed in many cancers (Adams and Cory 2007; Grobholz et al. 2002; Llambi and Green 2011; Mathupala et al. 2009), VDAC1 can be considered as a key protein in apoptosis regulation. These interactions can be prevented by VDAC1-based peptides that bind specifically to HK (Abu-Hamad et al. 2009; Abu-Hamad et al. 2008; Arzoine et al. 2009), Bcl-2 or Bcl-xL (Abu-Hamad et al. 2009; Arbel et al. 2012; Arbel and Shoshan-Barmatz 2010; Malia and Wagner 2007;), abolishing the cancer cell’s abilities to bypass the apoptotic pathway. Here, we focus on the interaction of VDAC1 with HK, Bcl-2 and Bcl-xL.
VDAC1-Hexokinase Interaction
Cancer cells rely on glycolysis as the main energy-generating pathway (i.e. the Warburg effect) and as a source of building blocks for proteins, nucleotides and lipids (Hanahan and Weinberg 2011; Mathupala et al. 2009). The metabolic re-programming of cancer cells includes marked over-expression of the mitochondrial-bound HK-I and HK-II isoforms (Grobholz et al. 2002; Mathupala et al. 2009), enzymes considered as rate-limiting for glycolysis and serving as the biochemical gate of this pathway (Mathupala et al. 2006; Shoshan-Barmatz et al. 2010a).
By binding to VDAC1 (Abu-Hamad et al. 2009; Abu-Hamad et al. 2008; Arzoine et al. 2009; Zaid et al. 2005), HK provides both a metabolic benefit and apoptosis-suppressive capacity that offers the cell a proliferative advantage while increasing its resistance to chemotherapy. The VDAC1-bound HK complex facilitates and promotes the high glycolytic tumor phenotype. HK, by association with VDAC1, gains direct access to mitochondrial ATP, reaching VDAC1 via the ANT in the IMM, allowing it to phosphorylate and ‘trap’ any incoming glucose (Pedersen 2008). With this direct coupling of mitochondrially generated ATP to incoming glucose via VDAC1-bound HK, mitochondria regulate glycolytic flux with that of the TCA cycle and ATP synthase to balance the energy requirements of the tumor cell with the biochemical requirements for metabolites (i.e. the anaplerotic and cataplerotic pathways, respectively) or metabolic precursors that are required by the tumor (Mathupala et al. 2006; Shoshan-Barmatz et al. 2010a, b). Thus, both the glycolytic pathway and other seminal metabolic pathways, like the pentose phosphate shunt, are regulated via the energy-coupling resulting from the formation of a VDAC1-HK complex. Moreover, the up-regulation of HK expression in tumor cells (Grobholz et al. 2002; Mathupala et al. 2009) and its binding to VDAC1 provide both a metabolic benefit and apoptosis-suppressive capacity that offers the cell a growth advantage and increases its resistance to chemotherapy (Arzoine et al. 2009). As part of a system that impacts cell growth, the VDAC1-HK complex thus offers a remarkable target for cancer therapy (see below).
VDAC1 Interaction with Bcl-2 Family Proteins
One of the hallmarks of cancer cells is their resistance to apoptosis through the development of a variety of strategies, including quenching of the mitochondrial apoptotic pathway by over-expression of anti-apoptotic proteins of the Bcl-2 family that are associated with resistance of tumors to chemotherapy (Adams and Cory 2007; Llambi and Green 2011). These apoptosis regulator proteins comprise both pro- and anti-apoptotic members and are classified by sequence homology based on the presence of up to four α-helical domains, termed Bcl-2 homology (BH) 1–4 domains (Adams and Cory 2007).
The mechanisms by which Bcl-2 family proteins regulate apoptosis are still not fully understood, yet it is well established that their activities are mediated via interactions with the mitochondria and controlling permeability of the OMM to Cyto c and other apoptotic factors (Adams and Cory 2007; Llambi and Green 2011). Moreover, accumulated evidence suggests that Bcl-2 family members act via interaction with VDAC1 (Abu-Hamad et al. 2009; Arbel et al. 2012; Arbel and Shoshan-Barmatz 2010; Malia and Wagner 2007; Shimizu et al. 2000; Sugiyama et al. 2002; Tsujimoto and Shimizu 2002; Yamagata et al. 2009). VDAC1 has been shown to interact with the Bax/Bak complex, Bcl-2 and Bcl-xL, as well as with Bax and Bim. Bax was found to increase VDAC1 conductance in one (Banerjee and Ghosh 2004), although not in a second study (Rostovtseva et al. 2004). The involvement of VDAC1 in Bax-mediated apoptosis has been proposed based on findings that in VDAC1-depleted cells, cisplatin-induced conformational activation of Bax was inhibited (Tajeddine et al. 2008) and because anti-VDAC antibodies inhibited Bax- and Bim-induced release of Cyto c (Shimizu et al. 2001). Moreover, it was shown that Bcl-xL can modify the VDAC1 oligomeric state by shifting the equilibrium from a monomeric to a dimeric state (Malia and Wagner 2007). Taken together, these data indicate that both the pro- and anti-apoptotic activities of Bcl-2 family proteins are mediated via interactions with VDAC1. Hence, interfering with these interactions should facilitate apoptosis induction and enhance the therapeutic effects of chemotherapeutic agents, like cisplatin. Such an approach involving VDAC1-based peptides interacting with Bcl-2 (Arbel and Shoshan-Barmatz 2010) and Bcl-xL (Arbel et al. 2012) has been applied to prevent the anti-apoptotic activities of these proteins, as considered below (Sect. 1.7).
16.1.6.2 HK, Bcl-2 and Bcl-xL Interact with the VDAC1 N-Terminal Domain
The N-terminal domain of the VDAC1 protein was shown to be accessible to anti-VDAC1 antibodies raised against this domain (Abu-Hamad et al. 2006). The same domain is exposed to kinases, as threonine-13 undergoes phosphorylation (Distler et al. 2007). Furthermore, the N-terminal domain of VDAC1 acts as a recruiting site for HK1, Bcl-2 and Bcl-xL and thus is a key structural feature mediating VDAC1 interaction with anti-apoptotic proteins and enabling their function (Abu-Hamad et al. 2009; Arbel et al. 2012; Arbel and Shoshan-Barmatz 2010; Arzoine et al. 2009; Malia and Wagner 2007; Shimizu et al. 2000; Sugiyama et al. 2002; Tsujimoto and Shimizu 2002), suggesting the exposure of this VDAC1 region outside the pore. Indeed, it has been further demonstrated the N-terminal region of VDAC1 is loosely attached to the barrel wall and can undergo translocation out of the pore (Geula et al. 2012a). Such movement of the N-terminal domain allows VDAC1 to interact with the anti-apoptotic proteins, HK, Bcl-xL and Bcl-2 (Abu-Hamad et al. 2009; Arbel et al. 2012; Arbel and Shoshan-Barmatz 2010). Thus, a peptide corresponding to the VDAC1-N-terminal domain can be used as a decoy to compete with native VDAC1 for interactions with HK, Bcl-2 and Bcl-xL, thereby preventing their anti-apoptotic activities.
Related posts:
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Pro-senescence Therapy for Cancer: Time for the Clinic
Pro-senescence Therapy for Cancer: Time for the Clinic
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


