Method
Measurement
Advantages
Disadvantages
Value in drug development decisions
Fasting serum insulin and/or C-peptidea
Serum insulin and/or C-peptide measured after an overnight fast
Fasting insulin; homeostasis model assessment (HOMA-B) is a mathematical index calculated from insulin and glucose concentrations
Simple; relatively inexpensive
Provides no information about stimulated insulin secretion; relatively high day-to-day variability
Relatively limited; of value as an exploratory endpoint requiring confirmation with a dynamic test of β-cell function
Tests of Stimulated Insulin Secretion
Method | Measurement | Advantages | Disadvantages | Value in drug development decisions | |
|---|---|---|---|---|---|
75 g oral glucose tolerance test | Insulin and/or C-peptide responses to 75 g oral glucose | Area under the curve for insulin; insulinogenic index (Δinsulin/Δglucose at t = 30 min) | Assesses β-cell response to the principal dietary secretagogue under well-standardized and internationally accepted conditions; extensive scientific literature against which to reference data; assesses integrated response that includes the effect of incretin hormones | Relatively high intra-individual and between-individual variability; β-cell response to secretagogues other than glucose not assessed; may be affected by gastric emptying rate | Similar to mixed meal tolerance test but less comprehensive assessment since restricted to glucose-stimulated insulin secretion; provides additional categorical data on glucose tolerance, i.e. progression/regression of diabetes |
Mixed meal tolerance test | Serum insulin and/or C-peptide to a meal of defined composition | Area under the curve for insulin; insulinogenic index (Δinsulin/Δglucose at t = 30 min) | Provides data relevant to normal physiology; flexible, i.e. nutrient components can be adjusted; assesses integrity of incretin axis | Relatively high intra-individual and between-individual variability; affected by gastric emptying rate | Provides dynamic data of physiological relevance; sensitive techniques for preliminary assessment of drug effects on insulin secretion; incretin axis is integral to responses |
Intravenous glucose tolerance test | Insulin and C-peptide responses to an intravenous bolus of glucose | Acute insulin response (AIR) | Provides dynamic data; widely used in clinical metabolic research | Relevance to normal physiology uncertain; does not generate dose–response data; incretin axis not assessed | May be a useful indicator of drug-induced effects on insulin secretion |
Hyperglycaemic clamp | Serum insulin and C-peptide responses to sustained hyperglycaemia generated by constant infusion of hypertonic glucose | First- and second-phase insulin responses to hyperglycaemia | Provides standardized assessment of insulin secretion | Insulin secretion is assessed in an unphysiological state of sustained hyperglycaemia; does not provide dose–response data; incretin axis not assessed | Moderately useful technique for assessing drug effects on insulin secretion |
Arginine potentiation test | Intravenous infusion of arginine superimposed on hyperglycaemic clamp | Acute insulin response to arginine (AIRmax) usually in the setting of controlled hyperglycaemia | Provides an assessment of near-maximal insulin secretion | Of limited physiological relevance; incretin axis not assessed | Of limited application in the context of drug development |
Graded glucose infusion | Serum insulin and C-peptide responses to a stepped incremental intravenous glucose infusion | Insulin secretion dose–response vs. glucose concentration; displacement of curve to left indicative of improved β-cell function | Provides detailed insulin-glucose dose–response data over range of glycaemia relevant to normal physiology as well as the pathophysiology of glucose intolerance and type 2 diabetes | Incretin axis not assessed; lengthy and labour intensive | Sensitive and reproducible technique that can provide robust data about drug-induced changes in insulin secretion, albeit without information about the incretin axis |
Conclusions
The range of different techniques for the assessment of insulin secretion and response to therapeutic interventions reflects the absence of a true reference method for drug development. No single method adequately captures all potentially relevant aspects of β-cell function. Selection of the most appropriate tests should be guided by the putative mechanism of action, e.g. direct secretagogue and activation of the incretin axis. A plurality of methods may be required to provide a comprehensive picture sufficient for making drug development decisions. Assessing the integrity of the incretin axis has come to prominence in recent years.
Introduction
Insulin is the principal anabolic hormone of the body and is essential for life [1]. The hormone exerts wide-ranging effects on carbohydrate, lipid and protein metabolism and regulates aspects of cell growth and differentiation [2]. Human insulin is composed of 51 amino acids and has a molecular weight of 5.8 kDa. The insulin molecule is a dimer comprised of an A-chain and a B-chain. The insulin gene is located on the short arm of chromosome 11 [3]. Insulin is synthesized within the β-cells of the pancreatic islets as a 110-amino-acid precursor. Preproinsulin is cleaved to proinsulin, and the latter is processed to insulin and C-peptide by specific endopeptidases and processed in the endoplasmic reticulum [4]. After passage through the Golgi apparatus, insulin is stored as hexamers in secretory granules awaiting release [5]. Insulin has a compact three-dimensional structure consisting of three helices and three conserved disulphide bridges [6]. This conformation is important for binding to and activating the insulin receptor. In the presence of zinc, the flux of which is regulated by zinc transporter proteins [7], and at a pH of approximately 6.0, insulin monomers assemble into higher order hexameric conformations [8]. The concentration of insulin within the storage granules of the β-cells is approximately 40 mmol/L [9]. The synthesis of insulin is regulated at both the transcriptional and translational level. Glucose metabolism within the β-cells stimulates the secretion of preformed insulin. Glucose is also the major physiological stimulator of the insulin gene [10].
Islet Physiology
The human pancreas contains approximately one million islets that comprise 1–2 % of the total mass of the gland. Islets contain four different endocrine cell types:
β-cells that produce insulin and constitute ~60–80 % of the endocrine cell mass
α-cells (~10–20 %) that produce glucagon
δ-cells (~5 %) that produce somatostatin
PP cells (<1 %) that produce pancreatic polypeptide
In order that β-cells can respond rapidly to prevailing blood glucose concentrations, islets receive approximately tenfold the blood flow of the surrounding exocrine cells of the pancreas. The capillaries surrounding islets have numerous fenestrations that facilitate nutrient exchange [11]. Heterogeneity in β-cell numbers between islets may be relevant to insulin secretion [12]. The activity of β-cells is synchronized both within and across islets. Endogenous insulin secretion is characterized by a pulsatile secretion with a periodicity of approximately 5 min and higher order swings [13, 14]. Alterations in the pulsatile secretion of insulin have been reported in patients with type 2 diabetes [15, 16] and also in glucose-intolerant first-degree relatives [17]. The increase in β-cell insulin secretion following ingestion of a meal normally signals a decrease or maintenance of glucagon secretion [18]. In the setting of deficient insulin secretion, relative hyperglucagonaemia contributes to hyperglycaemia in diabetes [18]. The higher proportion of α-cells to β-cell in the islets of some patients with type 2 diabetes has been attributed to a decrease in β-cell number rather than an increase in α-cell number [19]. Somatostatin, the pulsatility of which is aligned with insulin [20], inhibits the secretion of both insulin and glucagon [21].
Glucose-Stimulated Insulin Secretion
In health, insulin is secreted to precisely meet metabolic demand and maintain circulating glucose concentration within strict limits. The rates of hepatic glucose production and glucose utilization are regulated by circulating insulin levels, remaining closely matched and relatively constant from day to day. Endogenous insulin secretion is stimulated primarily by increases in blood glucose levels. Non-glucose initiators of insulin secretion include certain monosaccharides, amino acids and fatty acids [11, 22]. It has been suggested that insulin secretion may also be influenced by other factors acting directly via cell surface receptors [23]. The discovery of G-protein fatty acid receptors that are either directly (GPR40) or indirectly (e.g. GPR120) involved in insulin secretion has generated new targets for diabetes drug development (see below) [24].
Glucose is transported from the blood into the β-cell via the high-capacity glucose transporter (GLUT2) system. Inside the cells, it is metabolized by glucokinase to generate glucose-6-phosphate [25]. Glycolytic and oxidative metabolism of glucose elevates the cytosolic adenosine triphosphate/adenosine diphosphate (ATP/ADP) ratio. This closes ATP-sensitive potassium channels in the cell membrane. The resulting depolarization triggers opening of voltage-gated calcium channels. Increased intracellular calcium concentration allows the fusion of insulin-containing granules with plasma membrane and the subsequent release of stored insulin through interactions with Ca2+-sensitive proteins (Fig. 2.1).
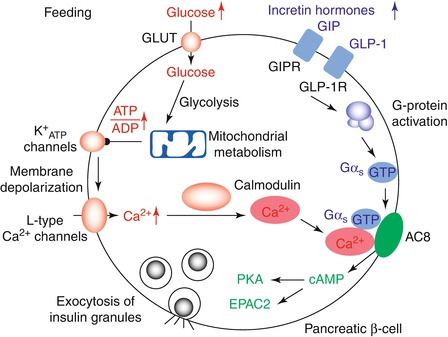
Fig. 2.1
Insulin secretion after a meal is typically initiated by a rise in extracellular glucose, which is detected by a metabolic signalling pathway. Aerobic glycolysis and mitochondrial oxidation produce metabolic signals, such as a rise in the ATP to ADP concentration ratio. This closes K+ATP-channels, depolarizes the plasma membrane and causes calcium influx that stimulates exocytosis. At the same time, the incretin hormones GLP-1 and GIP bind to their receptors, which are highly expressed on human pancreatic β-cells. This causes the activation of G proteins. The glucose and GLP1 signalling pathways converge on AC8, an isoform of adenylate cyclase that is abundant in pancreatic β-cells and fully activated when both Gαs–GTP and calcium–calmodulin are bound. The subsequent rise in cellular cAMP triggers exocytosis by protein kinase A (PKA) and EPAC2 (also known as RAPGEF4) (Reprinted by permission from β-cells Macmillan Publishers Ltd: Nature Cell Biology, Integrating insulin secretion and ER stress in pancreatic β-cells, K Lemaire and F Schuit, copyright 2012)
Initiators of insulin secretion – These induce insulin secretion both by triggering, i.e. involving closure of the KATP channels, and amplifying effects.
Potentiators of insulin secretion – The incretin hormone glucagon-like peptide (GLP)-1 (see below) and arginine do not initiate insulin secretion by themselves, but enhance insulin secretion in the presence of an initiator, e.g. glucose.
Even in the unstimulated (basal) state, insulin secretion is continually tuned by numerous stimulatory and inhibitory signals [26]. Insulin secretion is influenced by central neural activity [27] and paracrine factors [28, 29]. Autocrine regulation of insulin secretion has been proposed [30, 31], although the physiological relevance of such a mechanism in humans has been questioned [32].
Glucose-stimulated insulin secretion to a rapid and sustained increment in plasma glucose is characteristically biphasic (Fig. 2.2) [33]. A transient first phase of insulin secretion with an early peak is followed by a gradually developing and more prolonged second phase. It has been suggested that the first phase may represent release of insulin from rapidly mobilized storage granules, triggered by the influx of Ca2+; the second phase of insulin secretion may reflect ATP-dependent recruitment and release of granules from a reserve pool [5, 34]. The relevance of first-phase insulin secretion to glucose homeostasis in human physiology remains uncertain [35].
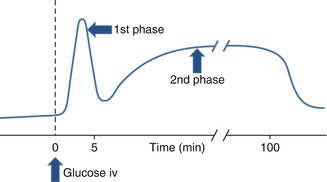
Fig. 2.2
Glucose-induced insulin secretion. A rapid first phase is followed by a slower and more sustained second phase of insulin release. iv intravenous. The biphasic response is observed in response to acute sustained experimental hyperglycaemia as induced by the hyperglycaemic clamp technique. See DeFronzo et al. [33] for methodological details
Role of the Incretin System in Insulin Secretion
At matched plasma glucose concentrations, orally ingested glucose elicits a substantially greater insulin secretory response than intravenous glucose; this is the so-called incretin effect [36]. The main incretin hormones, GLP-1 and glucose-dependent insulinotropic polypeptide (GIP), are released from intestinal endocrine cells (L cells and K cells, respectively) within minutes of nutrient ingestion. The potentiation of glucose-stimulated insulin secretion by the incretin hormones may account for approximately 50–70 % of postprandial insulin secretion [37, 38]. GLP-1 and GIP circulating in the blood are rapidly inactivated via enzymatic cleavage by dipeptidyl peptidase 4 (DPP-4) [39, 40]. GLP-1 and GIP act via specific G-protein-coupled cellular receptors to raise intracellular levels of cyclic adenosine monophosphate (AMP) and inhibit ATP-sensitive K+ channels, actions which induce β-cell exocytosis of insulin (see Fig. 2.1) [41]. Incretin-mediated augmentation of insulin secretion and inhibition of glucagon secretion are dependent on ambient glucose concentrations [42]. In addition, GLP-1 reduces gastric emptying, increases satiety and reduces food intake [43] as well as stimulating the transcription of glucokinase and the GLUT 2 transporter gene [44].
Defects in incretin axis physiology are implicated in the pathogenesis of type 2 diabetes. Delineation of these defects has led to the development of important new classes of glucose-lowering drugs, i.e. the DPP-4 inhibitors and GLP-1 analogues [40]. Reported reductions in GLP-1 responses in patients with type 2 diabetes provided the theoretical basis for these incretin-based glucose-lowering drugs [45]. The impact of hyperglycaemia and incretin effect on insulin secretion and β-cell function continues to be unravelled [46, 47]. In a placebo-controlled study of subjects (n = 22) with impaired fasting glucose, sitagliptin increased intact circulating GLP-1 concentrations but had no effect on postprandial plasma glucose, insulin or C-peptide concentrations [48]. While the secretion of GIP is near normal in patients with type 2 diabetes, the effect of this incretin on insulin secretion, particularly the late phase, is impaired [38].
Hepatic Clearance of Insulin
Insulin and C-peptide are co-secreted into blood that is directed to the portal vein, and insulin directly regulates hepatic glucose metabolism [16]. Hepatic first-pass clearance of insulin removes >50 % of insulin ensuring that the portal:systemic insulin gradient is 2:1 or higher. Insulin clearance, of which hepatic extraction is the main determinant, may be altered in common metabolic diseases [49] and possibly by certain glucose-lowering drugs [50]. Since it is not cleared by the liver, the serum concentration of C-peptide in the peripheral circulation provides a more reliable indicator of endogenous insulin secretion. The most robust assessment of dynamic β-cell insulin secretion would require blood samples drawn directly from the hepatic portal vein. Such a highly invasive approach is not feasible in clinical research, although the technique has been applied in nonhuman primate studies [51]. Prehepatic insulin secretion may be estimated by mathematical deconvolution of serum C-peptide in peripheral blood (see below) [52]. The half-life of serum C-peptide is longer than that of insulin (~20–30 vs. ~3–5 min). During dynamic tests of insulin secretion, the longer half-life in the circulation of serum C-peptide requires that blood sampling time points for measurement of C-peptide be adjusted relative to those for insulin. Impaired renal function reduces C-peptide clearance [53].
β-Cell Dysfunction in the Pathogenesis of Diabetes
Glucose metabolism is regulated via a feedback loop between islet β-cells and insulin-sensitive target tissues [54]. Within this physiological regulatory system, the sensitivity of the liver and muscle cells to insulin modulates the β-cell response [55]. When insulin resistance develops, usually due to combinations of genetic and lifestyle factors, β-cells maintain normal glucose tolerance via an appropriate increase in endogenous insulin secretion [56]. According to a widely accepted model of the pathogenesis of type 2 diabetes, hyperglycaemia results when sufficient amounts of insulin cannot be secreted to fully compensate for insulin resistance (see below) [55, 57]. In this scenario, the relative contributions of decreased β-cell mass versus decreased β-cell function remain uncertain [58]. In humans, β-cell mass increases through cellular proliferation during the first decade of life before stabilizing during adolescence [59]. In adulthood, hyperplasia and hypertrophy permit a degree of plasticity in compensation for insulin resistance [60]. If issues of sensitivity and precision can be resolved [61], quantitative imaging techniques, e.g. fluorescent exendin-4 derivatives [61, 62], may eventually complement conventional metabolic approaches for estimating β-cell mass [63]. Type 1 diabetes and type 2 diabetes are both characterized by progressive β-cell destruction (see below) in which apoptosis (programmed cell death) predominates. Since a low rate of β-cell turnover may continue throughout the human lifespan, β-cell number will fall if the rate of apoptosis or dedifferentiation is increased relative to neogenesis [64, 65].
Type 1 Diabetes
Selective immune destruction of β-cells is the principal pathogenic defect of type 1 diabetes [66]. The resulting insulin deficiency necessitates lifelong replacement therapy with subcutaneous administration of insulin or insulin analogues (or a successful pancreas or islet transplant). However, recent data suggest that even patients with type 1 diabetes of long duration may have low levels of residual insulin secretion [67, 68].
In clinical trials of patients with type 1 diabetes, β-cell function is commonly assessed by the serum C-peptide response to intravenous glucagon [69] or a mixed meal [70]. Glucagon is injected as an intravenous bolus with timed measurements of serum C-peptide [69]. The glucagon stimulation test may also be of value in clinical practice when therapeutic decisions may be are informed by assessment of endogenous insulin secretion [71].
Type 2 Diabetes
Islet β-cell insufficiency is central to the development and progression of type 2 diabetes. A decline in β-cell function antedates and predicts the onset of clinical diabetes [61]. Genetic and environmental factors are strongly implicated [55]. Characterization of defective insulin secretion in the precursor states of impaired fasting glucose (IFG) and impaired glucose tolerance (IGT) has provided valuable insights into the pathogenesis of type 2 diabetes [72]. In the United Kingdom Prospective Diabetes Study (UKPDS), β-cell function, determined using a modelling method (HOMA-B, see below), was reduced by approximately 50 % in middle-aged subjects at the time of diagnosis [73]. This is in line with estimates based on autopsies place the reduction of β-cell mass at approximately 40–65 % at the time of clinical presentation of type 2 diabetes [74]. A reduction in functional β-cell mass is evident during the long preclinical phase of type 2 diabetes. In parallel, the regulation of blood glucose both postprandially and in the fasting state deteriorates through stages of glucose intolerance before rising to levels diagnostic of diabetes. Insulin secretion rates rise through the range between normal glucose tolerance to IGT and decline thereafter [75]. Longitudinal studies have demonstrated progressive and variable, but not inevitable, loss of β-cell function in patients with type 2 diabetes [76, 77]. As the number of β-cells per islet falls, plaques of amyloid fibrils, which may contribute to β-cell loss, are deposited [78, 79]. Hyperglycaemia (glucotoxicity), both acute [80] and chronic [81], and prolonged elevations of plasma concentration of nonesterified (‘free’) fatty acid concentrations (lipotoxicity) [82] are detrimental to β-cell function and viability [10, 83]. Oxidative and endoplasmic reticulum stress together with inflammation have been identified as pathways to β-cell loss that are potentially amenable to targeted therapeutic interventions [84]. As hyperglycaemia develops, insulin pulses are attenuated [85] and circulating levels of proinsulin and partially processed proinsulin products become are elevated, albeit remaining at low concentrations relative to insulin [86]. Loss of acute glucose-stimulated insulin secretion is accompanied by alterations in β-cell phenotype and in gene and protein expression.
Uncommon monogenic forms of diabetes in which β-cell defects have been well delineated, e.g. forms of maturity-onset diabetes of the young (MODY) [87, 88], contrast with the complexity of type 2 diabetes in which the relative contributions of genetic, epigenetic and nongenetic factors to β-cell failure remain uncertain [61, 89]. To date, the utility of molecular genetics in identifying personalized therapeutic approaches for diabetes has been confined to relatively small subgroups of patients. Diabetes due to hepatic nuclear factor-1α mutations is especially sensitive to sulphonylureas [90]. Neonatal diabetes due to rare activating mutations in genes encoding the ATP-sensitive potassium channel subunits Kir6.2 or SUR1 can be successfully treated with high-dose sulphonylureas [90].
Strategies to Improve β-Cell Function for the Treatment and Prevention of Diabetes
Type 1 Diabetes
The causes of type 1 are complex and multifactorial, involving genetic and environmental factors [91]. The natural history of type 1 diabetes involves a prolonged and variable latent preclinical period that culminates in the destruction of pancreatic β-cells [92]. When autoimmune-mediated β-cell loss reaches a critical point, hyperglycaemia develops. A temporary partial recovery of β-cell function may occur after initiation of insulin therapy – the so-called honeymoon period – before waning again thereafter. A diverse range of strategies for averting type 1 diabetes (primary prevention) or preserving β-cell function in subjects with recently diagnosed type 1 diabetes (secondary prevention) have been explored in clinical trials [93]. Examples of interventions directed at arresting the immune process include nicotinamide, insulin injections, oral insulin, nasal insulin, glutamic acid decarboxylase, cyclosporine, teplizumab and abatacept [93]. These challenging intervention studies have been aided by the development of biomarkers of autoimmunity [94] and serial tests of β-cell function [95]. The latter include the mixed meal tolerance test (see below) and glucagon-stimulated serum C-peptide response; these methods should not be assumed to provide equivalent results [96]. Recent research has shown that endogenous glucagon dynamics are also altered early in the course of type 1 diabetes with elevated levels in response to a mixed meal challenge [97]. The plurality of aforementioned therapeutic targets attests to the complex pathophysiology of β-cell attrition in type 1 diabetes. Novel non-immunomodulatory strategies are being explored. For example, preclinical evidence suggests that verapamil, a calcium channel blocker used for the treatment of hypertension and cardiac arrhythmias, may enhance β-cell survival and function [98]. In a recent study of patients with newly diagnosed type 1 diabetes the lipid-modifying agent atorvastatin appeared to slow the decline of β-cell function (stimulated C-peptide secretion), in a subgroup defined by higher levels of inflammation-associated immune mediators [99].
Type 2 Diabetes
In obese subjects with glucose intolerance behavioural interventions that indirectly improve insulin secretion reduce the risk of progression to diabetes [100]. Many drugs have been shown to increase insulin secretion in vitro, but few have therapeutic potential [101]. Thiazolidinediones, which do not directly stimulate insulin secretion, have also been shown to improve β-cell function and reduce the risk of new-onset diabetes in insulin-resistant women with a history of gestational diabetes [102]. The latter observation illustrates the pathophysiological relevance of the feedback loop between insulin secretion and insulin action [103]. Lifestyle modification is the cornerstone of controlling hyperglycaemia in established type 2 diabetes [104]. Bariatric surgery procedures [105] or voluntary dietary calorie restriction of a sufficient degree can rapidly lower blood glucose concentrations and improve β-cell function [106]. A doubling of β-cell function in obese patients with type 2 diabetes 1 year after gastric bypass surgery has been reported [107]. When standard behavioural changes prove insufficient, pharmacotherapy is required to control hyperglycaemia. Sulphonylureas have the mainstay of pharmacological strategies to enhance insulin secretion for decades [108]. Agents in this class act primarily by blocking ATP-sensitive potassium channels in the β-cells thereby stimulating insulin release [109]. Dipeptidyl peptidase (DPP)-4 inhibitors, which potentiate insulin secretion in a glucose-dependent fashion by reducing the breakdown of GLP-1, have superior safety and tolerability profiles to sulphonylureas [110]. Unlike sulphonylureas, which continue to stimulate insulin secretion even during hypoglycaemia [111], DPP-4 inhibitors and GLP-1 analogues promote insulin exocytosis only when blood glucose levels are elevated. This property reduces the risk of hypoglycaemia when these drugs are used as monotherapy or in combination with other agents that have a low propensity to cause hypoglycaemia [112, 113]. Preclinical data suggesting effects of DPP-4 inhibitors on functional β-cell mass and pancreatic insulin content in rodent models raised hopes that the natural history of type 2 diabetes might be modified [114]. The UKPDS demonstrated an inexorable loss of β-cell function with sulphonylureas and insulin [73]. As yet, however, there is no firm evidence from clinical studies that GLP-1 agonists [115] or DPP-4 inhibitors [114] have durable effects on β-cell function. Progressive loss of β-cell function is regarded as the main reason that many patients with type 2 diabetes ultimately require insulin replacement therapy [73]. The hypothesis that β-cell function may be preserved by using specific combinations of glucose-lowering drugs is being tested in clinical trials [116]. New drugs designed to improve β-cell function that have entered clinical trials in recent years include:
DDP-4 inhibitors (including agents requiring only once-weekly dosing) [117]
Bile acid sequestrants with indirect effects on incretin secretion [120]
GPR40 agonists [121]
GPR119 agonists [122]
Glucokinase activators [124]
Interleukin-1 β antagonists [125] (Table 2.1)
Table 2.1
Current and future pharmaceutical agents with direct or indirect actions on islet β-cells for the treatment of diabetes
Primary pharmacological effect on β-cellsa
Insulinotropic agents
Sulphonylureasb
Direct: closure of KATP channels; stimulation of insulin secretion
Meglitinides
Direct: closure of KATP channels; stimulation of insulin secretion
DPP-4 inhibitors
Indirect: elevation of GLP-1 levels; potentiation of insulin secretion
Incretin mimeticsc
Direct: stimulation of GLP-1 receptors; potentiation of insulin secretion
Insulin sensitizers
Metformin
Indirect: reduction in insulin resistance; elevation of GLP-1 levels
Thiazolidinediones
Indirect: reduction in insulin resistance; reduction in lipotoxicity
Glucose-lowering drugs with non-insulin-dependent actions
α-glucosidase inhibitors
Indirect: reduction in glucotoxicity
SGLT-2 inhibitors
Indirect: reduction in glucotoxicity
Insulin; insulin analogues
Indirect: reduction in glucotoxicity
Potential agents and/or targets for future treatment of type 2 diabetes
Bile acid sequestrants
Indirect: stimulation of intestinal GLP-1 secretion
GPR40 agonists
Direct: activation of FFA1 receptor
GPR119 agonists
Direct and indirect: activation of GPR119 receptor; activation of incretin axis
Dual GLP-1/GIP agonists
Direct: stimulation of GLP-1 and GIP receptors
Glucokinase activators
Direct: activation of glucose-sensing enzyme glucokinase
IL-1β antagonist
Direct: reduced intracellular cytokine signalling
HDL lipoproteinsd
Direct: calcium-dependent insulin secretion
In parallel, potential cardioprotective actions of GLP-1, highly attractive in the context of the enhanced risk of cardiovascular disease in type 2 diabetes, have also been explored [126]. A novel hypothesis that extends to the broader cardiometabolic risk profile of patients with type 2 diabetes is that strategies to raise high-density lipoprotein (HDL) levels may improve β-cell function [127]. The cholesteryl ester transfer protein inhibitor torcetrapib improved glycaemic control in atorvastatin-treated patients with type 2 diabetes. However, it remains to be determined whether this effect was a direct consequence of raising HDL levels [128]. Further elucidation of the molecular regulation of β-cell mass and function offers the prospect of additional therapeutic agents to preserve insulin secretion [129].
Other Drugs with Effects on β-Cell Function
In the design of studies of novel therapies for diabetes, the potential for confounding by drugs that have effects on β-cell function should be carefully considered (Tables 2.1 and 2.2). In studies of new pharmacological entities for type 2 diabetes, individuals who are treatment naïve or on stable metformin monotherapy are preferred (see Chap. 9). Washing off sulphonylureas and DPP-4 inhibitors is a routine practice in early-phase studies of diabetes drugs although careful monitoring of glycaemic control is required according to regulatory guidelines [130]. When thiazolidinediones [131] or GLP-1 analogues [132] are temporarily withdrawn, a sufficient length of time is required to ensure that the any metabolic effects of these drugs have fully dissipated [130].
Table 2.2
Selected drugs commonly used in the treatment of disorders other than diabetes with reported direct effects on β-cell function
Effect on insulin secretion | |
|---|---|
Anti-hypertensive drugs | |
β-adrenergic blockersa | Decreased |
Angiotensin-converting enzyme inhibitors | Increased |
Angiotensin receptor | Increased |
Calcium channel blockersb,c | Decreased |
Imidazoline agonists | Increased |
Diazoxideb | Decreased |
Lipid-modifying drugs | |
Statinsd | Decreased |
Antipsychotic drugs | |
Second-generation antipsychotic agentsd | Decreased |
Calcineurin inhibitors | |
Cyclosporine, tacrolimus | Decreased |
Somatostatin receptor agonists b | |
Octreotide, lanreotide | Decreased |
Aside from glucose-lowering medications, many patients with diabetes are treated with antihypertensive and lipid-modifying drugs that may have class-specific effects to either increase or decrease endogenous insulin secretion [133, 134]. Recent reports of adverse effects of statins on glucose metabolism are pertinent in this context [135]. Certain statins may affect insulin secretion through direct and/or indirect effects on β-cell calcium channels [136].
Clinical Research Methods for Quantifying β-Cell Function
No consensus exists on a reference method for assessing endogenous insulin secretion in humans [103]. It has been argued that conclusions concerning insulin secretion and the impact of therapeutic interventions may vary according to the use of different methodologies [137]. Each of the commonly used methods focuses on a defined facet of β-cell function [138]. Assessment of β-cell function may be undertaken either in the basal, i.e. non-stimulated state, or following challenge with various secretagogues, either in isolation or combination [139]. The administered stimuli may be either physiological, i.e. a test meal of known macronutrient composition, or pharmacological, e.g. intravenous injection of glucose or arginine.
The advent of the therapeutic era of incretin mimetics has focused attention on the incretin axis and broader aspects of islet function. The effect of DPP-4 inhibitors on insulin secretion is indirect, mediated through improvements in incretin axis signalling [140]. Comprehensive evaluation of a novel incretin mimetic requires assessment of both the incretin axis and the resulting effect on insulin secretion [37]. Approximately 30–50 % of circulating GLP-1 in the postprandial state consists of active GLP-1, the rest being the inactive truncated fragment [141]. During treatment with DPP-4 inhibitors, the relative proportion of the active form of the hormone, i.e. GLP-17-36, is increased [142]. DPP-4 inhibitors preferentially lower postprandial glucose, with less marked effects on fasting hyperglycaemia [110]. Metformin [143] and α-glucosidase inhibitors [144] have also been shown to increase circulating GLP-1 levels. Part of the glucose-lowering effect of incretin mimetics is mediated via reductions in glucagon secretion [145–147]. Clinical methods for the assessment of α-cell function are not as sophisticated as those directed towards β-cell function [72]. The glucagon receptor has been identified as a therapeutic target for type 2 diabetes [148]. However, toxicity issues including elevations in hepatic transaminases direc and plasma lipids have been reported with glucagon receptor antagonists [148].
Non-stimulated (Basal) Assessments
Fasting Insulin and/or C-Peptide
The feedback loop between the β-cells and insulin-responsive tissues requires that circulating insulin concentrations be interpreted in the context of the prevailing blood glucose level. Changes in both insulin sensitivity and β-cell function are evident as glucose tolerance deteriorates [149]. When fasting hyperglycaemia is present, the predicted level of hyperinsulinaemia would be higher if β-cells were able to fully overcome any defect in insulin action through compensatory increased insulin secretion [150]. Demonstration of an effect on residual β-cell function is an important consideration in studies of new drug therapies for type 2 diabetes. This is most readily accomplished by measurement of serum or plasma C-peptide after an overnight fast. Several classes of diabetes drugs either stimulate insulin secretion (e.g. sulphonylureas, DPP-4 inhibitors) or require the presence of sufficient insulin to exert their glucose-lowering effects (e.g. metformin, thiazolidinediones) [108]. Exceptions include α-glucosidase inhibitors [151] and sodium glucose cotransporter 2 (SGLT2) inhibitors [152].
Proinsulin
The use of insulin-specific immunoradiometric assays eliminates cross-reactivity with proinsulin and partially processed proinsulin molecules [153]. Loss of the normal pulsatile oscillations in insulin secretion [85] and increased ratios of circulating proinsulin to insulin [153, 154] are regarded as markers of compromised β-cell function in prediabetic states of glucose intolerance. In patients with type 2 diabetes, the ratio of proinsulin to insulin is inversely related to β-cell function [155]. In studies of an insulin secretagogue and an insulin sensitizer in patients with type 2 diabetes, a differential effect of the two classes of glucose-lowering drugs was observed on plasma proinsulin and the proinsulin to immunoreactive insulin ratio [155]. Glibenclamide (glyburide) increased, whereas rosiglitazone decreased, the concentration of proinsulin and the proinsulin-to-insulin ratio [155]. In a 12-month head-to-head study of these two drugs, rosiglitazone reduced the concentrations of insulin, proinsulin and split proinsulin compared to glibenclamide [156]. As expected, rosiglitazone improved insulin sensitivity, whereas glibenclamide therapy was associated with worsening insulin resistance [156]. These results were replicated in ADOPT (A Diabetes Outcome Progression Trial) [157]. Over the 4-year course of ADOPT, rosiglitazone had more favourable effects on β-cell function than glibenclamide [158]. The improvements in β-cell function with rosiglitazone, which were accompanied by a decrease in insulin resistance, were associated with a more durable effect on glycaemic control [157, 158]. Whether the beneficial effects of rosiglitazone on β-cell function were indirect via improved insulin sensitivity or resulted from a direct β-cell action [159] could not be determined from this study (see below). Metformin, which was also studied in ADOPT, had effects on β-cell function and glycaemic control that were intermediate to those observed with rosiglitazone and glibenclamide [157, 158].
HOMA-B
Homeostasis model assessment is a method for assessing insulin secretory capacity from fasting plasma insulin and glucose concentrations. HOMA-B has been widely applied in clinical metabolic research, including diabetes drug development.
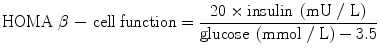 An early study of diabetic and nondiabetic subjects reported a coefficient of variation (CV) for HOMA-B measured on two occasions of approximately 30–40 % [160]. Subsequent studies using specific insulin assays in larger samples have yielded CVs closer to 10 % [138]. The use of triplicate samples may improve the CV with smaller sample sizes [138]. In comparative studies, HOMA-B has been shown to correlate with measures of insulin secretion obtained using the hyperglycaemic clamp (see below) in diabetic (r = 0.59) and nondiabetic subjects (r = 0.71) [160]. Since HOMA values are rarely normally distributed, the data should be logarithmically transformed and presented as geometric means with appropriate measures of dispersion.
An early study of diabetic and nondiabetic subjects reported a coefficient of variation (CV) for HOMA-B measured on two occasions of approximately 30–40 % [160]. Subsequent studies using specific insulin assays in larger samples have yielded CVs closer to 10 % [138]. The use of triplicate samples may improve the CV with smaller sample sizes [138]. In comparative studies, HOMA-B has been shown to correlate with measures of insulin secretion obtained using the hyperglycaemic clamp (see below) in diabetic (r = 0.59) and nondiabetic subjects (r = 0.71) [160]. Since HOMA values are rarely normally distributed, the data should be logarithmically transformed and presented as geometric means with appropriate measures of dispersion.
The HOMA model apportions the basal state of insulin and glucose in terms of insulin resistance and β-cell function [138]. Insulin sensitivity and insulin secretion are mutually related as a hyperbolic function in which insulin resistance is compensated by increased insulin secretion [54, 103]. Reporting HOMA-B data in isolation, i.e. without considering the prevailing level of insulin sensitivity for the individual, could lead to erroneous conclusions about β-cell function [103, 138]. The validity of assumptions inherent in HOMA-B has been challenged [161]. It has been argued that measures of β-cell function and insulin sensitivity should be as independent as possible; clearly, HOMA does not satisfy this requirement [103]. HOMA-B is a measure of β-cell activity rather than health; accordingly, the temporary improvement observed in response to a classic secretagogue, i.e. a sulphonylurea, should be regarded as reflecting the mechanism of action of the drug rather than any intrinsic change in functional capacity [73].
HOMA-B provides an index of insulin secretory capacity in the non-stimulated state at serum insulin levels primarily of relevance to hepatic glucose regulation [162]. This focus may detract from the ability of HOMA-B to quantify defective β-cell function relative to dynamic tests. In the Insulin Resistance Atherosclerosis Study (IRAS), HOMA-B underestimated the magnitude of the β-cell defect in subjects with IGT and newly diagnosed diabetes compared with the acute insulin response assessed using an intravenous glucose challenge (see below) [163]. In a study in which insulin secretion was compared with pancreatic histology, the glucose-stimulated C-peptide-to-glucose ratio appeared to predict β-cell area better than HOMA [164]. With these caveats in mind, HOMA-B offers a simple and useful approach for assessing β-cell function that can be of value in evaluating the actions of new diabetes drugs. For example, the insulin sensitizer pioglitazone lowers blood glucose in patients with type 2 diabetes primarily by improving insulin action [131]. Thiazolidinediones also improve β-cell function through indirect mechanisms, an effect demonstrable as an increase in HOMA-B [165]. For DDP-4 inhibitors, which primarily enhance postprandial insulin secretion, an oral glucose challenge would be more logical than an assessment of fasting β-cell function. However, in a recent meta-analysis of placebo-controlled clinical trials of sitagliptin, improved β-cell function was evident as an increased HOMA-B index [166]. Note that HOMA-B cannot be used in patients treated with exogenous insulin.
Dynamic Tests of β-Cell Function
Oral Glucose Tolerance Test
The 75 g oral glucose tolerance test (OGTT) is the reference method for diagnosing diabetes and other categories of glucose dysregulation [167]. Thus, regression from one diagnostic category to another, e.g. IGT to normal glucose tolerance, in response to therapy can be determined. For example, in the DREAM (Diabetes Reduction Assessment with Ramipril and Rosiglitazone Medication) study in adults with IFG and/or IGT, rosiglitazone significantly reduced the incidence of type 2 diabetes and increased the likelihood of regression to normoglycaemia compared to placebo [168].
The OGTT can also provide an integrated assessment to the β-cell response to an intestinal glucose stimulus that includes activation of the incretin axis. As discussed above, the incretin effect on the islets includes enhancement of glucose-stimulated insulin secretion in concert with reduced glucagon release [169]. When assessing the response to therapeutic agents whose primary mode of action is mediated via the incretin axis, e.g. DDP-4 inhibitors, the response of the major incretin hormones (GLP-1, GIP) can be quantified and related to the stimulation of insulin secretion and suppression of glucagon secretion. For example, in a placebo-controlled crossover study performed in patients with type 2 diabetes, a single oral dose of sitagliptin (25 mg or 200 mg) dose-dependently inhibited plasma DPP-4 activity over 24 h [170]. Sitagliptin increased active levels of GLP-1 and GIP, increased serum insulin and C-peptide levels, decreased plasma glucagon levels and reduced the rise in blood glucose during a 75 g OGTT [170].
For paired comparisons, the area under the curve (AUC) for serum insulin (or C-peptide) offers a simple exploratory assessment of the change in endogenous insulin secretion in response to a therapeutic intervention that incorporates early and later phases of insulin release. Consideration should be given to the methods used for data analysis in this scenario [171]. Since the OGTT is not standardized in terms of attained blood glucose concentrations, methods for normalizing insulin secretion to the glucose stimulus have been developed. A number of empirical indexes of modelling approaches have been proposed which relate insulin responses to increments in blood glucose. For example, the insulinogenic index, which reportedly correlates with first-phase insulin release assessed using an intravenous glucose bolus, is calculated as the ratio of the increment of insulin to glucose above baseline 30 min (Δinsulin/Δglucose) after an oral glucose challenge [172]. In ADOPT [157] (see above), the insulinogenic index was used to study the β-cell responses to rosiglitazone, glibenclamide and metformin. A higher acute change in the insulinogenic index was observed over the first 6 months with glibenclamide than with rosiglitazone or metformin [158]. Thereafter, glibenclamide was associated with a significantly faster rate of decline in insulinogenic index versus rosiglitazone (11.1 vs. 6.0 % per year), the decline with metformin being intermediate. Despite the fact that glibenclamide is a classic insulin secretagogue, the mean insulinogenic index with glibenclamide was lower than those in the other treatment groups beyond 2 years [158]. Patients who failed monotherapy (with any agent) had a lower median insulinogenic index at baseline compared to those who completed the 4-year trial [158]. High within-subject variability for the insulinogenic index has been reported [173] which should be considered in the design of studies with smaller sample sizes.
Related posts:
 Regulatory Considerations for Early Clinical Development of Drugs for Diabetes, Obesity, and Cardiometabolic Disorders
Regulatory Considerations for Early Clinical Development of Drugs for Diabetes, Obesity, and Cardiometabolic Disorders
 Early Phase Metabolic Research with Reference to Special Populations
Early Phase Metabolic Research with Reference to Special Populations
 Omics: Potential Role in Early-Phase Drug Development
Omics: Potential Role in Early-Phase Drug Development
 Measurement of Energy Expenditure
Measurement of Energy Expenditure
 Assessment of Body Composition
Assessment of Body Composition
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


