Fig. 40.1
Definition and assessment of T cell functional avidity and TCR-pMHC binding affinity/avidity. (a) T cell functional avidity is a biological readout that reflects T cell responsiveness upon antigen-specific stimulation and is assessed by quantifying T cell responses (i.e., cytotoxic activity, IFN-γ production or proliferation) when exposed to titrated doses of antigenic peptide. EC50, defined as the peptide concentration producing half-maximal response. (b) TCR-pMHC binding affinity and avidity are biophysical readouts that describe the strength of monomeric/multimeric binding interaction(s) between the TCRs and their antigens and are typically measured using surface plasmon resonance (SPR) and pMHC multimers, respectively. Adapted from [31]
CD8 T cell functional avidity is predominantly guided by the strength of TCR binding and kinetic interactions to antigenic peptides presented by MHC class I molecules (i.e., pMHC). Importantly, the binding and kinetic attributes of this interaction are determinant parameters that influence almost every aspect of T cell biology, including thymic selection (reviewed in [26]), differentiation into effector and/or memory T cells ([27], reviewed in [28]), and functional efficacy (reviewed in [29–31]), and may offer superior metrics to evaluate the quality of T cell responses. However, due to technical limitations, the assessment of TCR-pMHC binding avidity or kinetics is still infrequently determined in research or patient immunomonitoring or in the selection of tumor-infiltrating T cells used for adoptive cell therapy. TCR-pMHC binding interactions can be measured in terms of affinity or avidity (Fig. 40.1b), both of which can directly impact the overall functional T cell response. The TCR binding affinity refers to the physical strength by which a single TCR binds to a single pMHC complex and is typically measured by surface plasmon resonance (SPR). Conversely, the TCR avidity describes the binding strength of TCR-pMHC interactions when assessed in the cellular context, includes the contribution of the co-receptors (e.g., CD8), and has often been estimated using recombinant soluble pMHC multimers. TCR-pMHC binding affinity/avidity is inversely proportional to the dissociation equilibrium constant K D, defined as the ratio of the dissociation rate (i.e., k off) and association rate (i.e., k on) under equilibrium conditions (reviewed in [29–31]).
40.3 Tumor-Specific T Cell Responses Are Mediated by TCRs of Low Binding Affinity/Avidity
Peripheral T cell repertoire diversity is achieved through random somatic gene segment rearrangements of the TCR alpha and beta chains during thymic development. The TCR repertoire is then shaped by positive intrathymic selection events allowing generating a peripheral T cell pool that can interact with the host pMHC complexes. However, to limit the development and/or activation of potentially harmful self-reactive T cells, the ones displaying TCRs of relative high affinity/avidity toward self-pMHC are deleted or anergized by mechanisms of central (also defined as negative thymic selection) and/or peripheral tolerance (reviewed in [32, 33]). As such it is now commonly accepted that the peripheral T cell repertoires targeting self versus nonself antigens may vary dramatically in terms of TCR-pMHC binding affinity/avidity (Fig. 40.2).
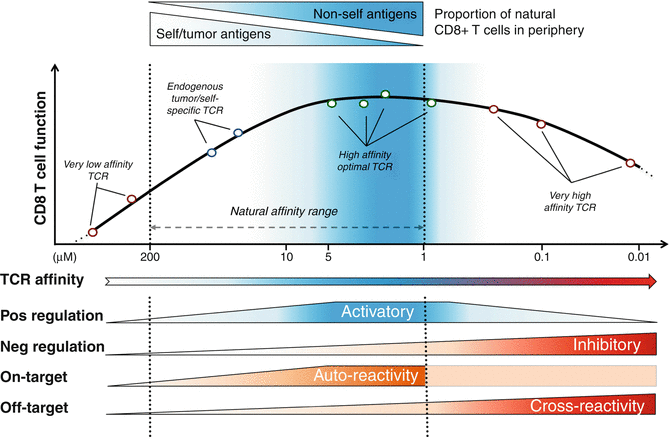
Fig. 40.2
Model integrating the relationship between T cell function, TCR affinity, and positive/negative regulators modulating cell responsiveness. A panel of CD8 T lymphocytes engineered to express antitumoral TCRs with incremental affinities toward the HLA-A2/NY-ESO-1 epitope (x-axis) was assessed for functional capacity (y-axis). An optimal window of TCR-pMHC affinity can be detected in the upper natural range (K D from 5 to 1 μM), corresponding to the range where most peripheral nonself-/virus-specific T cells can be detected [36]. We recently described how positive regulators of T cell function including CD28 and TNFR cofactors are enriched in T cells lying within the optimal affinity window and how the inhibitory receptor PD-1 and SHP-1 phosphatase are involved in restricting T cell activation and responsiveness in TCR-engineered CD8 T cells of very high supraphysiological affinities [47]. On-target toxicity is known to follow the functional and activatory signatures of CD8 T cells, while off-target responsiveness and toxicity are described to increase along TCR-pMHC affinity, within the supranatural affinity range (reviewed in [58]). Adapted from [48]
Many tumor-associated antigens that are shared among cancer patients, such as overexpressed (e.g., Her2/neu, WT1, or telomerase), differentiation (e.g., Melan-A/MART-1, gp100 or tyrosinase), cancer-testis (e.g., NY-ESO-1 or MAGEs), or oncofetal (e.g., AFP or CEA) antigens, are not truly cancer specific but are also expressed by healthy tissues (reviewed in [34, 35]). As a consequence, the natural T cell repertoire targeting self/tumor antigens is thought to express TCRs of relative low affinity/avidity, whereas high-avidity T cells are very rare since they are eliminated through mechanisms of central and/or peripheral tolerance. Aleksic and colleagues [36] demonstrated that the K D values of these interactions are typically in the range of 200 μM to 10 μM (with the mean around 100 μM), while virus-specific TCRs interact with K D ranging down to 1 μM (with the mean value around 10 μM). Nonetheless, these tolerance mechanisms spare T cells that can react to self/tumor antigens with relative low TCR-pMHC binding affinity/avidity [37–40]. Thus, although cancer patients may have many self-/tumor-specific T cells, their TCR affinity/avidity remains relatively low and in fact eventually too low to mediate an effective antitumor immunity. Therefore, increasing the TCR-pMHC affinity and/or avidity of tumor-specific T cells is of particular interest in the context of cancer immunotherapies.
40.4 Optimization of TCR-pMHC Binding Affinity/Avidity Against Cancer Cells
Adoptive transfer of TCR-engineered T cells is a recent type of immunotherapy, which aims to establish and boost immune reactivity toward poorly immunogenic tumors. This strategy relies on the optimization of the TCR sequence to increase its affinity/avidity for cognate tumor antigens with the aim to adoptively transfer them back to patients. Indeed, adoptive cell transfer of engineered T cells augments the functional and protective capacity of tumor-antigen reactive CD8 T cells [41, 42]. During recent years, we established a unique panel of human CD8 T cells engineered with TCRs of progressive increasing affinities against the tumor antigen NY-ESO-1 presented in the context of the HLA-A2, obtained through structure-based rational predictions [43, 44]. We observed that T cells expressing TCRs with affinities in the upper natural range (K D from 5 to 1 μM) displayed greater biological responses when compared to those expressing intermediate affinity wild-type TCR (K D at 21.4 μM) or very low affinity (K D > 100 μM) ( [45, 46]; Fig. 40.2). Strikingly, further increase within the supraphysiological TCR affinity range (K D < 1 μM) led to drastic functional decline, with impairment in global gene expression, signaling, and surface expression of activatory/costimulatory receptors [46, 47]. Major findings revealed that maximal T cell effectiveness was limited by at least two mechanisms (Fig. 40.2). First, we observed the preferential expression of the inhibitory receptor PD-1 within T cells expressing very high TCR affinities, and this correlated in those cells with functional recovery upon PD-1 ligand 1 (PD-L1) blockade [47]. Second, contrasting to PD-1 expression, we observed a gradual up-regulation of SHP-1 phosphatase in CD8 T cells with increasing TCR affinities. Consequently, pharmacological inhibition allowed further incremental gaining of cell function in all engineered T cells, according to their TCR-binding affinities [47]. Our observations fit nicely with other studies performed both in mice and human models (reviewed in [48]) and provide strong evidence that T cell activation and signaling can be increased up to a given affinity threshold for the TCR-pMHC interaction and that above this threshold, T cells may not develop productive functions.
The importance of TCR-pMHC binding parameters has also been demonstrated in several clinical trials (reviewed in [49–52]) (Table 40.1). Initial reports have shown that in contrast to T cells of low avidity, high-avidity tumor-specific T cell responses were often associated with autoimmunity [53–55]. On-target reactivity was more recently observed in a clinical trial whereby melanoma patients received autologous blood mononuclear cells transduced with affinity-optimized TCRs against the differentiation tumor antigen Melan-A/MART-1 ([41]; Table 40.1). Compared to the native low TCR avidity (referred as DMF4), the DMF5 TCR of higher binding avidity toward HLA-A2/Melan-A showed improved clinical efficacy. Importantly, T lymphocytes expressing increased avidity self-/tumor-specific TCRs also targeted normal tissues expressing the cognate antigen (e.g., melanocytes in the skin, eye, and ear for both Melan-A and gp-100-specific T cells), thus mounting harmful cytotoxic immune responses in vivo [41]. Similar on-target reactivity was observed when using a TCR engineered against the human carcinoembryonic antigen (CEA) in patients with refractory metastatic colon carcinoma [56]. Whereas all three patients experienced decline in serum CEA levels, severe transient inflammatory colitis was further reported in those patients [56]. Interestingly, studies based on the A*0201/NY-ESO-1 cancer-testis antigen model showed that genetically optimized T cells were those displaying maximal functionality, leading to objective clinical responses, but without in vivo cross-reactivity or major adverse events [42, 57]. These apparently contradictory results could be explained by the differences in tissue distribution of NY-ESO-1 antigen being only expressed in testis cells when compared to Melan-A or CEA antigens, both widely found in normal melanocytes or epithelial cells of the gastrointestinal tract, respectively. Consequently, the choice of antigen specificity for adoptive cell transfer of affinity-improved T cells is of crucial importance (reviewed in [52]).
Table 40.1
Clinical trials performed with T cells of optimized TCR binding affinity/avidity
Type of cancer | Targeted antigen | TCR origin (name) | ORa,b (%) | CRb,c (%) | On-/off-target toxicity (%)b | Reference | |
|---|---|---|---|---|---|---|---|
Melanoma | A*0201/ Melan-A | Human (DMF5) | 6/20 (30) | 0/20 (0) | 8/20 (40) | On-target: vitiligo, uveitis, and hearing loss | [41] |
Melanoma | A*0201/ gp100 | Mouse (154) | 3/16 (19) | 0/16 (0) | 1/16 (6) | On-target: vitiligo, uveitis, and hearing loss | [41] |
Colorectal carcinoma | A*0201/ CEA | Mouse | 1/3 (33) | 0/3 (33) | 3/3 (100) | On-target: colitis | [56] |
Melanoma and synovial sarcoma | A*0201/ NY-ESO-1 | Human (1G4) | 22/38 (58) | 5/38 (13) | 0/38 (0) | NAd | |
Melanoma and multiple myeloma | A*01/ MAGE-A3 | Human (3A3) | 0/2 (0) | 0/2 (0) | 2/2 (100) 2 deaths | Off-target: cardiogenic shock | |
Melanoma, synovial sarcoma, and esophageal cancer | A*0201/ MAGE-A3 | Mouse (118AT) | 5/9 (55) | 1/9 (11) | 4/9 (44) 2 deaths | Off-target: mental status changes, seizure, and coma | [65] |
Melanoma | A*0201/ Melan-A | Human (DMF5) | 0/14 (0) | 0/14 (0) | 2/14 (14) | AEe | [105] |
Multiple myeloma | A*0201/ NY-ESO-1 | Human (NY-ESOc259) | 16/20 (80) | 14/20 (70) | 0/20 (0) | NAd | [106] |
Another parameter to be considered is the risk of increased cross-reactivity to structurally related self-peptides, resulting in off-target toxicities (reviewed in [58]; Table 40.1). Using experimental models, it has been demonstrated that T cells, whose TCR binds to pMHC complexes with very high avidities (K D < 1 nM) lose antigen specificity and can become cross-reactive [59–61]. Similarly, critical results from recent clinical trials revealed that affinity-enhanced TCRs engineered above a certain threshold could lead to severe off-target side effects in patients (reviewed in [62]). Patients treated with TCRs engineered for enhanced affinity toward the cancer-testis HLA-A1/MAGE-A3 tumor antigen developed off-target recognition toward a similar but not identical peptide from the cardiac muscle-specific protein titin [63], resulting in a serious adverse effect (SAE) and fatal toxicity against cardiac tissue [64]. Moreover, two patients infused with autologous anti-HLA-A2/MAGE-A3 TCR-engineered T cells experienced mental changes, leading to neurologic toxicities and death, possibly due to cross-reactivity toward the MAGE-A12 self-antigen expressed in the human brain [65].
These results demonstrate not only the functional potency of affinity-improved TCRs with substantial antitumor effector functions in vivo but also highlight the potential safety concerns for those TCR-engineered T cells (reviewed in [49–52, 58, 62]) (Table 40.1). Altogether, we and others propose that the rational design of improved self-specific TCRs for adoptive T cell therapy may not need to be optimized beyond the natural TCR affinity range to achieve optimal T cell function and avoidance of unpredictable risk of cross-reactivity (reviewed in [48, 66]; Fig. 40.2). Thus, there is an urgent need to withstand for better preclinical evaluations allowing assessing for precise antigen specificity of the engineered TCRs and identifying the best suitable tumor antigens for adoptive cell transfer with affinity-improved TCRs. Importantly, TCR optimization through affinity alteration must include the evaluation of optimal T cell responsiveness and lack of on-target and off-target side effects due to self-reactivity to ensure the safety of TCR-engineered T cells in future clinical trials (reviewed in [49–52, 58, 62]). In that regard, new strategies are required, allowing for the identification and selection of those naturally occurring but rare self/tumor antigen-specific T lymphocytes of highest TCR binding avidities and functional capacities within the physiological TCR affinity range, and will be further described thereafter.
40.5 TCR-pMHC Binding and Kinetic Measurements
Several technologies have been employed to characterize in great detail the TCR-pMHC binding affinity/avidity and to dissect the kinetic contributions of association k on and dissociation k off rates to such molecular interactions. In this section, we will summarize the different methods currently used, focusing on their respective advantages and disadvantages.
Early comparative crystallography analysis revealed a lack of correlation between TCR-pMHC structure and T cell function, suggesting that T cell activation must rely on dynamic processes [67]. Quantitative measurements of the kinetic and binding parameters of TCR-pMHC interactions were thus undertaken, mostly using SPR biosensors. This technology allows for the simultaneous detection of association and dissociation kinetic rates as well as the molecular affinity (1/K D), providing the so-called “3D affinity” parameters, since one of the two molecules is flowed in solution across a sensor chip, on which the potential interacting target is attached (Fig. 40.1b). Data collected from SPR studies revealed that natural human TCR-pMHC binding interactions were of relative weak affinities (K D ranging from 500 μM to 1 μM) compared to other biomolecular interactions, with rapid off- and slow on-rates [68–70]. Yet, an inherent caveat of the SPR technology is that it requires the laborious and time-consuming production of soluble TCR and pMHC molecules and ignores the binding contribution of CD8 co-receptors and/or other membrane molecules to the overall TCR-pMHC interaction. Recently, new generations of imaging technologies, coupling microscopy to SPR affinity measurements [71, 72], as well as quartz crystal microbalance (QCM) biosensors [73, 74] have been engineered to allow label-free detection of real-time interactions and kinetics between membrane proteins directly at the surface of living cells. These new technologies could potentially be exploited in the context of low affinity TCR-pMHC interactions at the surface of living self/tumor antigen-specific T cells.
Besides SPR, other technologies based on mechanical or fluorescent assays have also been recently developed that enable the deduction of k on and k off kinetics directly at the interface between a living T cell and a surrogate APC, or between a T cell and a supported planar lipid bilayer. As such, these surface-based membrane interactions are designed as “2D interactions” [75–77]. 2D analyses showed good correlations with T cell activation and function, but revealed faster k on and k off kinetics than those found with 3D SPR technology [78–80]. Although 2D approaches allow for the measurements of TCR-pMHC binding parameters in a more physiological way than the 3D technology, both approaches should be viewed as highly complementary. Indeed, 2D analyses require specialized equipment and are time consuming, precluding for the rapid and high-throughput screen of living antigen-specific T cells that could be useful for adoptive cell immunotherapy (reviewed in [81, 82]). It should be noted that a very recent study revealed the successful measurement of single-cell 2D TCR affinity and subsequent TCR sequencing directly from human primary CD8 T cells [83], opening the way to the rapid selection of individual therapeutic TCRs for adoptive cell transfer immunotherapy.
Alternative technologies based on the direct detection and rapid analysis of live antigen-specific CD8 T cells led in the early 1990s to the development of soluble pMHC monomers [84]. However, the low affinity characterizing TCR binding to pMHC precluded any direct detection of antigen-specific CD8 T cells by flow cytometry and/or microscopy using these soluble pMHC monomers [85]. Increasing the molecular valency and binding avidity of pMHC monomers through controlled multimerization (e.g., dimers, tetramers, decamers) was then performed, enabling direct binding of multimers on live antigen-specific T cells [86] and initiating a whole new area of T cell detection and analysis [81, 87]. Yet, multivalency favors TCR-pMHC clustering and augments the likelihood that a dissociating pMHC rapidly rebinds to another juxtaposed TCR. As such, evaluation of TCR-pMHC affinity using multimer-based saturation binding experiments, as well as association and dissociation kinetic assays, generated biased results, which often did not correlate to the monomeric SPR-based affinity and kinetic data [88]. Consequently, the brightness of multimer staining to cell membrane of TCRs (i.e., the mean fluorescence intensity or MFI), which reflects the total binding capacity of the multimeric molecules at equilibrium, failed to consistently correlate with in vitro functional activity or in vivo protection [20, 89–91].
A major technological improvement was achieved with the development of reversible multimers (reviewed in [31]). Reversible multimers are structurally similar to multimers, but they are engineered in such a way that pMHC monomers can be disrupted from the multimeric scaffold upon addition of a stimulus. Reversible Streptamers consist of a fluorescent scaffold molecule (StrepTactin) coupled to several pMHC monomers carrying a streptag, a linear peptide optimized to bind to StrepTactin [92]. D-biotin binds StrepTactin with higher affinity than streptag and is therefore able to compete for the same binding site, disrupting the multimeric complex and releasing the pMHC monomers at the cell surface. Since pMHC monomers do not stably bind to TCRs, they rapidly dissociate from the TCRs, allowing for the identification and isolation of practically “untouched” antigen-specific CD8 T cells, while preserving their phenotypical and functional status [93]. Based on the same principle, the reversible multimers called NTAmers are made of His-tagged pMHC linked to fluorescent streptavidin carrying an engineered nitrilotriacetic acid (NTA) linker [94] (Fig. 40.3a). Upon addition of imidazole at low and nontoxic concentration, the NTA complexes rapidly decay into pMHC monomers allowing FACS sorting of antigen-specific CD8 T cells without inducing adverse effects on the cell integrity such as activation-induced cell death [81, 94].
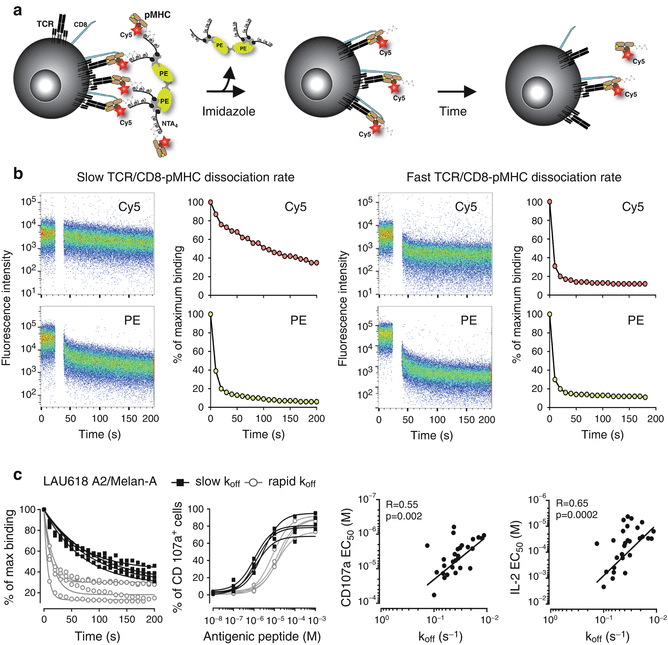
Fig. 40.3
NTAmer-based monomeric dissociation assay allows the identification of antitumor T cells of higher TCR-pMHC avidity and functional potency. (a) Schematic representation of the NTAmer-based monomeric dissociation assay. CD8 T cells are stably stained with multimeric NTAmers composed of streptavidin-PE (green)-NTA4 (gray) and peptide-MHC (brown) monomers containing Cy5-labeled β2m (red). Upon addition of imidazole, NTAmers rapidly decay in Cy5-labeled pMHC monomers (middle panel). Cy5-labeled pMHC monomers subsequently dissociate from cell-associated TCRs (black) and CD8 (blue) according to the intrinsic TCR/CD8-pMHC dissociation rate (k off) (right panel). Adapted from [31]. (b) Representative NTAmer-dissociation staining (left panels) and corresponding fitting curves (right panels) obtained from CD8 T cell clones defined as slow or fast TCR-pMHC dissociation rate (k off). Imidazole is added after 1 min of baseline recording (white gap), and dissociation curves are followed over time within the Cy5 (pMHC monomers) and PE (NTA4 scaffold) channels by flow cytometry. The corresponding monomeric dissociation rates (k off) are analyzed in Prism (GraphPad Software, Inc.). (c) Relationship between TCR-pMHC binding avidity and T cell functional avidity. Representative NTAmer-based monomeric dissociation curves (first panel from the left) and CD107a-based titration curves (second panel) obtained for HLA-A2/Melan-A-specific CD8 T cell clones isolated from a melanoma patient (LAU618) and arbitrarily separated into rapid (white circle) and slow (black squares) dissociation rates (k off). Correlations (Spearman coefficient r and P value) obtained between TCR-pMHC avidity (monomeric TCR-pMHC k off) and functional avidity (CD107a degranulation EC50, third panel, and IL-2 production EC50, fourth panel)
Thanks to the fluorescent labeling of the individual pMHC monomers contained in the reversible multimeric complexes, it has recently become possible to monitor and quantify the monomeric dissociation of pMHC from the TCRs directly at the surface of antigen-specific T cells [95–98]. Reversible two-color Streptamers successfully allowed determining monomeric dissociation kinetics of nonself, virus-specific TCRs on human and mouse T cells using a real-time microscopic-based strategy [95]. Specifically, Nauerth and coworkers [95] reported that virus-specific CD8 T cells with longer half-lives (low k off) exhibited increased functional avidity and better in vivo protective capacity than T cells of shorter half-lives (high k off). Moreover, the Streptamer technology allowed for the simultaneous measurement of k off on hundreds of antigen-specific CD8 T cells, which represents a great advantage since it is not limited to the generation of clonal T cell populations. However, the significant lag time required for the Streptamer to decay into monomeric pMHC molecules (around 60 s) as well as the photobleaching effect associated with the microscopic assay prevented the detection of rapid TCR-pMHC off-rates, which are typically found within the self-/tumor-specific CD8 T cell repertoires of lower TCR affinities. Recently, the Streptamer-based experimental settings were further optimized for k off measurements of ex vivo unsorted, polyclonal antigen-specific CD8 T cells using a conventional flow cytometer and may now become suited for high-throughput applications [98].
In parallel, our group used a two-color version of the reversible NTAmers [94] to assess TCR-pMHC dissociation kinetics directly on living CD8 T cells by flow cytometer [96] (Fig. 40.3a, b). The accuracy of the NTAmer approach was validated by finding strong correlations between NTAmer-based monomeric dissociation rates and those obtained by SPR measurements [96]. In contrast to the Streptamer technology, the rapid decay of the NTAmer scaffold into its pMHC monomeric constituents (2–3 s) made it possible to precisely analyze the dissociation kinetics of a wide spectrum of TCR affinities, with a special emphasis for self-/tumor-specific CD8 T cells, which are known to be of low affinities and rapid k off [96, 97] (Fig. 40.3c). Collectively, the two-color reversible multimer technology (i.e., Streptamer and NTAmer) enables the real-time quantification of monomeric TCR-pMHC dissociation kinetics directly at the surface of living primary CD8 T cells within large numbers of nonself/virus and self/tumor antigen-specific CD8 T cell clones. For the time being, no monomeric-based pMHC assays have emerged, that would allow the precise quantification of the k on association kinetics or the K D affinity directly on living T cells.
Related posts:
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


