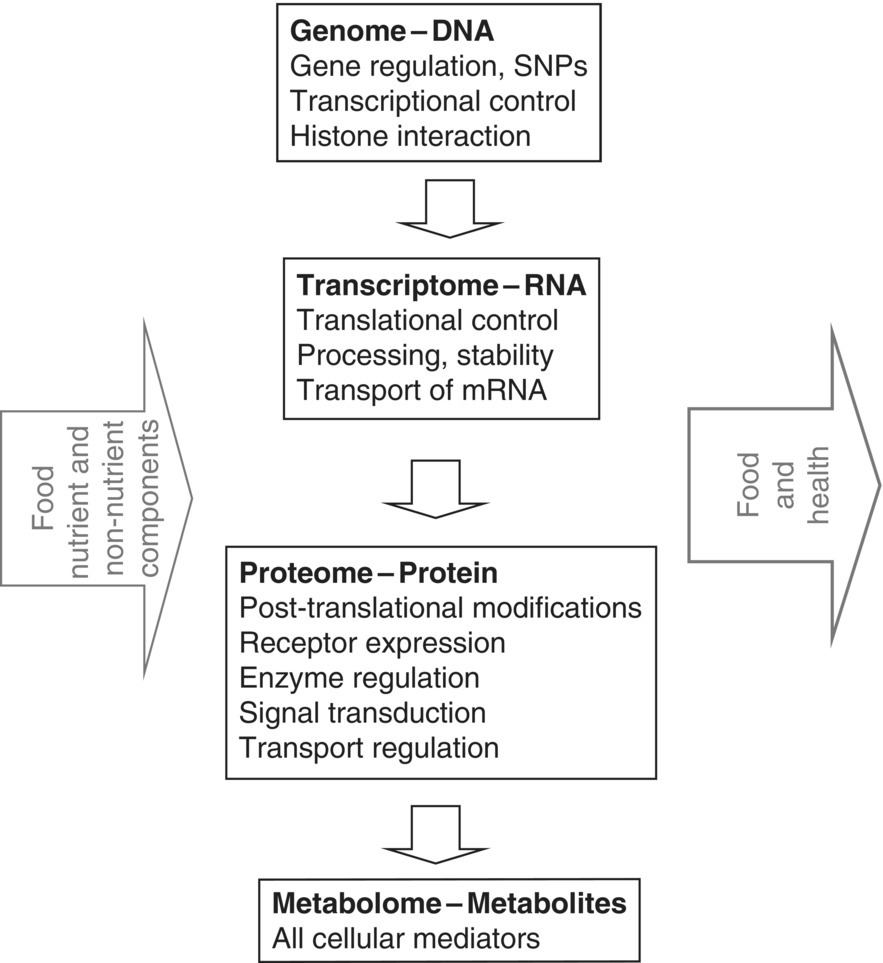
13 Helen M Roche,1 Baukje de Roos2 and Lorraine Brennan1 1 University College Dublin 2 University of Aberdeen An individual’s nutritional phenotype represents a complex interaction between the human genome and environmental factors during that individual’s lifetime. With the advent and continual advancement of state-of-the-art ‘omics’ technologies, it is now possible to investigate the dynamic, two-way interaction between nutrition and the human genome, which determines gene and/or protein expression and the consequent metabolic response, the combination of which is reflected in an individual’s health status. High-throughput molecular ‘omics’ technologies, defining the genome, epigenome, transcriptome, proteome and metabolome, are providing an unprecedented opportunity to advance our understanding of the molecular effects of nutrition on health and common diet-related diseases. This chapter will explain the basic principles relating to each technology platform, and thus explore the potential for high-throughput genomic, transcriptomic, proteomic and metabolomic technologies within human nutrition research. Every technology has great potential to advance the state of the art; however, it also needs to be acknowledged that each also has its limitations. It is important to note that these tools are best used in conjunction with strong nutritional/biochemical/physiological biomarkers of health and disease, with a view to using ‘omics’ technologies to gain a further understanding of the molecular processes wherein nutritional status and/or interventions contribute to that state. Genomics refers to gene-sequencing approaches to determine the total genetic information, or genome, of a cell or an organism. In very simple terms, genomics attempts to explore the deoxyribonucleic acid (DNA) sequence, or the precise order of the four bases – adenine, guanine, cytosine and thymine – in a nucleotide or strand of DNA within the genome. Genomics includes any method or technology that is used to determine the order; the common methods include genotyping arrays or ‘BeadChip’ technologies. The advent of very powerful genomic technologies has made it dramatically easier and very cheap to sequence DNA, so that it is now possible to determine complete genome sequences relatively easily. The expression of each gene when translated leads to the formation of a protein, which together with many other proteins that are coded by other genes forms tissues, organs and systems, and these combined constitute the whole organism. As illustrated in Figure 13.1, the flow of genetic information from the genome, transcriptome and proteome is reflected in the metabolome, and both nutrient and non-nutrient food components can interact at each level to affect the relationship between the human genome, nutrition and health. Figure 13.1 The health effects of nutrient and non-nutrient food components may be regulated via specific molecular interactions with the genome, transcriptome, proteome and/or metabolome. Adapted from Roche, H.M. (2006) Nutrigenomics – new approaches for human nutrition research. Journal of the Science of Food and Agriculture, 86, 1156–1163. Genetic polymorphisms are different forms of the same allele in the population. The ‘normal’ allele is known as the wild-type allele, whereas the variant is known as the polymorphic or mutant allele. A polymorphism differs from a mutation because it occurs in a population at a frequency greater than 1%. Alleles with frequencies less than 1% are considered as a recurrent mutation. The Human Genome Project demonstrated that the human genome is almost identical (99.9%) between individuals. The remaining 0.1% variation is principally accounted for by single nucleotide polymorphisms (SNPs). These common forms of inherited genetic variation involve a single base change in the DNA and account for almost 90% of variation between individuals. It is estimated that each of our genes contains approximately 10 variations or SNPs in its code from the standard gene. Important terminology related to genomics and nutrigenetics is presented in Box 13.1. It is very important to note that not all polymorphisms have a functional impact. SNPs can occur in both the coding and the, more abundant, non-coding regions of the human genome. In addition, a single base change in a coding region of a gene does not necessarily alter gene function or the resultant amino acid sequence. Functional SNPs are those that may alter the amino acid sequence or a transcription-factor binding element. DNA variants in several genes can interact with numerous environmental factors, including nutritional status, to determine several common, polygenic, diet-related diseases, including cardiovascular disease (CVD), obesity, type 2 diabetes (T2D), some cancers and so on. The research challenge lies in understanding how the combination genetic variation(s) determine cellular homeostasis, whole-body metabolism and health, because each SNP makes a relatively small contribution. This complexity is in stark contrast to the more unusual, but very profound, monogenic conditions such as phenylketonuria (PKU) or familial hypercholesterolaemia, which are attributable to a single genetic defect that interacts with nutritional status. In the case of PKU, the defective gene for the hepatic enzyme phenylalanine hydroxylase (PAH) interacts with the amino acid phenylalanine to cause disease unless that amino acid is removed from the diet to maintain health. More recently, copy number variation (CNV) has been identified as another common form of genetic variation. It is estimated that about 0.4% of the human genome differs with respect to CNV. As yet it has not been associated with susceptibility or resistance to diet-related diseases, but it is possible that this type of genetic variation may also be linked to nutrition and health. In addition, alterations in DNA structure without changes in the underlying gene, nucleotide or DNA sequence, referred to as epigenetics, can also have important functional effects. Epigenetic modifications alter gene expression due to mechanisms beyond DNA sequence modifications. The molecular basis of this is complex, but it involves modifications including DNA methylation, DNA acetylation and histone modification. These changes in DNA structure cause the activation/deactivation of gene expression, thereby affecting the subsequent transcriptome, proteome and metabolome (as illustrated in Figure 13.1) without changing the DNA sequence. Epigenetics will be dealt with in greater detail in Chapter 14, but briefly, DNA methylation refers to the addition of methyl groups to the CpG islands in the promoter regions of DNA and is associated with gene inactivation, which in turn affects transcriptional activity. Importantly, epigenetic modifications remain through cell division for the duration of the cells’ life and can also be passed on to future generations, a process known as transgenerational epigenetic inheritance of functional DNA variation, where the genes express themselves differently despite being identical from a sequence perspective. Changes in DNA methylation are a potential molecular mechanism through which diet and lifestyle interventions mediate their effects on the transcriptome. Initial research showed that global DNA hypermethylation, and/or conversely hypomethylation, was implicated in the development and progression of cancer. From the nutritional perspective, folate status can affect DNA methylation, which in turn can affect gene expression through mechanisms that are being actively researched. Within the context of type 2 diabetes, it has been shown that family history is associated with differences in the DNA methylation status of important cell signalling and metabolic genes in skeletal muscle and adipose tissue. Furthermore, the DNA methylation status is modifiable by diet and exercise interventions, wherein the methylation state may modify the degree to which DNA can be transcribed into ribonucleic acid (RNA), thus affecting the transcriptome, and subsequent proteome and metabolome. The term nutrigenetics focuses on investigating the relationship between common genetic variations or polymorphisms (or epigenetic modifications) and the nutritional environment. Such gene–nutrient combinations may determine an individual’s nutrient requirements, their metabolic response and/or their responsiveness to a nutritional intervention, all of which may predispose an individual to a lesser or greater risk of developing a diet-related disease. Personalised nutrition approaches sometimes focus on the effect of genetic variation in response to dietary change, because some polymorphisms or epigenetic states may determine an individual’s response and/or the therapeutic efficacy of a dietary intervention, which may in turn determine the outcome of certain disease states. The candidate gene approach has been traditional for identifying the genes involved in a diet-related condition. The candidate genes can be identified according to biological function and/or linkage studies. Each subject with and without the disease (case and control groups) donates a DNA sample, the sequence of which is determined using an appropriate sequencing or array platform. Then association tests are carried out for significant differences in the allele frequencies of the SNPs of interest. If one variant of the allele is more frequent in the ‘at-risk’ patient group compared to a healthy control population, then the SNP is ‘associated’ with the disease. This candidate gene approach can be used within a case-control and/or prospective study design. While there have been a number of studies published using this approach, and interesting research has shown different interactions between different-risk SNPs and nutritional status. Nevertheless, it is important to note that overall there has been limited success from candidate gene studies, in terms of defining the genetic determinants of diet-related diseases. This limitation reflects the fact that a number of metabolic pathways with several candidate genes are often involved in diet-related disease and metabolic responses. Furthermore, it is important to be aware of the potential limitations of gene association studies. Firstly, it is essential to replicate any potential finding in an independent cohort, as too often positive results in one cohort have not been replicated in subsequent studies. The main reasons for this are inadequate statistical power, multiple hypothesis testing, population stratification, publication bias and phenotypic differences. It is becoming increasingly evident that the identification of true genetic association in common multifactorial conditions, such as obesity, T2D or cancers, requires large studies consisting of thousands rather than hundreds of subjects. In addition, it is desirable to demonstrate a functional effect of a given SNP. For example, if a polymorphism introduces a missense change in the coding region of the peroxisome proliferator activator receptor gamma (PPARγ) gene, it probably represents a functional variant. Alternatively, functional assays may show greater or lesser activity of the protein arising from the gene. As an example, it has been shown that the Pro12Ala polymorphism in the transcription factor PPARγ gene was associated with less DNA-binding affinity and reduced transcriptional activity. Lastly, the absence of large single-gene effects and the detection of multiple small effects accentuate the need for the study of larger populations in order to identify reliably the size of the effect that we now expect for complex diseases. Positional cloning involves mapping the susceptibility/causative loci purely on their chromosomal location, using multigenerational pedigrees and/or a large number of sibling pairs. This allows identification of genes without any prior knowledge of biological function or the disease mechanism. Linkage and linkage disequilibrium (LD) analysis relies on the fact that genes with similar chromosome positions will only rarely be separated during genetic recombination, so that susceptibility to causative genes can be localised by search for genetic markers that co-segregate with disease. However, to date this approach has identified relatively few candidate genes relevant to diet-related diseases. One of the earliest significant linkage peaks was at chromosome 2q37.3, which led to the identification of Calpain 10 (CAPN10) as a new putative diabetes gene. The authors implicated an A to G polymorphism in intron 3 of the gene encoding CAPN10 with a greater risk of T2D. Subsequent to this initial report, several groups showed modest associations between CAPN10 polymorphisms and metabolic phenotypes associated with T2D. Nevertheless, other groups have failed to show a relationship between CAPN10 and metabolic traits indicative of T2D. The lack of a firm gene–phenotype relationship arising from a gene, identified using a positional cloning approach, may be due to a number of causes of inconsistency in association studies, including population-specific patterns of LD, population-specific environmental triggers and gene–gene or gene–nutrient interactions. Genome-wide association studies (GWAS) focus on determining the prevalence of several (thousands of) SNPs simultaneously. The principle is similar for the candidate gene approach, with GWAS comparing the DNA of groups of cases versus control subjects. The DNA sequence of the cases and controls is determined using SNP arrays to determine whether the frequency of gene variant(s) is greater in the population with the condition versus controls. The associated SNPs are then considered to mark a region of the human genome that influences disease risk. GWAS goes beyond the candidate gene approach, which is limited to one or a few genetic regions based on prior knowledge and research bias. GWAS investigates the entire genome, thus it has the opportunity to identify novel genetic determinants. Again this approach has limitations, as while GWAS can identify new SNPs in DNA that are associated with a disease, it cannot specify which genes are causal. The potential limitations of GWAS relate to optimal study design. Sufficient numbers of accurately phenotyped cases and controls, adequate measures to control and correct for multiple testing to avoid the generation of false positive results and control for population stratification are important design issues that need to be addressed at the outset of a new study. Next-generation sequencing (NGS) and high-throughput sequencing are technologies that have the capability greatly to enhance efficiency, producing thousands or millions of sequences at once. High-throughput sequencing technologies are intended to lower the cost of DNA sequencing compared to standard methods. NGS achieves high-throughput sequencing by completing the sequencing process of multiple DNA fragments that overlap in parallel or simultaneously. Then the final DNA sequence is reassembled by collating the DNA sequence information from the multiple overlapping DNA fragment sequences. Transcriptomic
Application of ‘Omics’ Technologies
13.1 An introduction to ‘omics’ technologies: Comprehensive tools to advance nutrition research
13.2 What is genomics and why is it an important technology?
Genomics: Experimental approaches to identify gene–nutrient interactions
13.3 Transcriptomics: Quantification of multiple-gene mRNA expression
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel


Full access? Get Clinical Tree


