Fig. 7.1
Overview of copy number and SNP microarray methodology. This figure summarizes the “wet lab” (a) and the analytic (b) tasks associated with DNA copy number and SNP microarray testing. (a) The “wet lab” steps will vary based on the platform utilized, but aspects of the methodology that are common to most platforms include the isolation of DNA from the specimen to be evaluated (this could be from frozen specimens or from FFPE specimens); labeling of the DNA (with SNP array methods differentially labeling the polymorphic forms [illustrated by a red or green label for the different polymorphic sequences]); denaturation of the specimen DNA to obtain single-stranded DNA; hybridization of the complementary sequences from the specimen to the array (with exact matches for the SNPs having a higher affinity for the array probe than mismatched sequences); washing of the array to remove nonspecific binding; scanning of the signals present on the array; and assessment of the intensity of each probe on the array. (b) The analysis protocol and software used will also vary from vendor to vendor, but follows the common logic of quantifying the probe signal intensity of the test sample compared to control samples (either real specimens or “in silico” specimens) for each site and ordering the information according to its chromosomal location. (i) For SNP arrays, the two types of polymorphic sequences are designated as either allele pattern “A” or allele pattern “B”. By assigning each allele an arbitrary numerical value (e.g., A = 0.5; B = −0.5), one can then calculate a SNP “score” for each locus and plot the data for each probe. In a balanced complement, both of the two chromosomes to which the sequence is localized might have the “A” form (value of 1.0) or the “B” form (value of −1.0); or one chromosome might have the “A” form while the second chromosome has the “B” form (value of 0.0). (ii) By plotting the SNP allele peak value on the Y axis for each probe, the latter of which are ordered on the X axis based on their map locations along the length of the chromosome (from the tip of the short arm to the tip of the long arm) the overall data can be visualized. For example, a balanced complement (two copies) would have an allele value of either 1.0, 0, or −1.0 at each probe. (iii) This process is reiterated for each of the thousands of SNP probes included in the array design. Given that the homozygous AA, BB or heterozygous AB allele patterns occur at approximately a random frequency, the allele peak pattern that emerges for a balanced (two copies) complement has three lines. Typically the centromeric regions of probes (as well as the short arm regions for the acrocentric chromosomes) are not included in the arrays (seen as a gap in the pattern) because the sequences localized to these regions are not unique to a single chromosome (and would hybridize to multiple chromosomes, thereby confounding the data interpretation)
Interpretation Methods
Copy Number Changes
As the generic phrase “copy number” implies, the data that results from a copy number microarray study provides information about the number of copies of characterized sequences that are present in the genome, with the queried sequences typically being spaced close enough together to allow for full genomic coverage (e.g., the average intergenic marker spacing for the Affymetrix CytoScan HD array is 1,737 base pairs [Affymetrix web site]). In addition to the “backbone” coverage, most arrays are also designed to have enrichment of probe coverage in genomic regions to ensure the detection of copy number variations in loci that are well recognized to have an association(s) with a health/developmental condition (e.g., the intragenic coverage for the Affymetrix CytoScan HD array is an average of 553 base pairs for cancer genes and 384 base pairs for genes associated with constitutional syndromes). Given the large size of the data resulting from these studies (thousands to millions of data points depending on the platform [e.g., the CytoScan HD array includes data for 2,696,550 copy number markers and 743,304 SNPs]), specialized software programs are used to assess the findings. The software programs will vary from vendor to vendor, but tend to have the common functionality of enabling the user to set criteria for designating regions that are indicative of a copy number gain or loss (e.g., cut-off levels). This designation is usually based on the number of contiguous (side-by-side) probes showing an alternation in copy number, as well as the size of the region demonstrating an alternation. In addition to size-related metrics, the software programs also provide information about the genomic location of the regions involved in the alternation (nucleotide coordinates) to enable the cytogeneticist or other specialist evaluating the data to compare each potential copy number variation with databases of known benign, uncertain, and clinically relevant changes (e.g., Database of Genomic Variants; International Collaboration for Clinical Genomics; Online Mendelian Inheritance of Man; UCSC Genome Browser, Ensembl, and others). Furthermore, the specialist(s) evaluating the data will often complete a current literature search using the gene(s) and/or nucleotide locations to enable her/him to make decisions regarding the clinical relevance of the observed alteration.
To allow one to visualize the thousands to millions of data points generated from the microarray study, the software programs provide graphical displays that integrate the probe data with information about their chromosomal location (and genomic database information). The copy number changes may be presented in a log ratio format, a direct copy number format, or a smoothed signal format (which incorporates a measure of the variation observed from contiguous probes). Given that this technology is based on comparisons of the amount of DNA present, it provides no direct information about the type of chromosomal alteration underlying the copy number alteration. Also, DNA copy number microarray studies will not detect truly balanced structural cytogenetic abnormalities since two copies of the chromosomal regions involved in the rearrangement would be present (albeit relocated to a different chromosomal location). This limitation of the technology is important since several solid tumors have pathognomonic balanced rearrangements. Thus, a full characterization of a solid tumor may best be completed from the use of more than one diagnostic assay (e.g., FISH and microarrays or PCR, or sequencing technologies and microarrays).
In addition to the copy number values, the SNP microarray platforms also provide data for the allele patterns. Briefly, as the name “single nucleotide polymorphism” implies, a SNP shows variation in the nucleotide sequence present for a particular region (Fig. 7.1). By definition, variations involving these regions are observed in at least 1 % of the population, but may occur in higher proportions (those with higher polymorphic indices can be more informative in microarrays). The two “forms” of these sequence polymorphisms can be designated as form “A” and “B” (Fig. 7.1). Given that a chromosomally normal person has two homologs for each of their autosomes (one they inherited from their mother and one they inherited from their father), their SNP pattern for any given autosomal locus could be either homozygous (AA or BB) or heterozygous (AB). For the sex chromosomes, the allele patterns will vary between males and females to reflect the presence of either two X chromosomes (females) or a single X and one Y chromosome (males). The SNP patterns present along the length of each chromosome can be represented as an “allele peak” or “B-allele frequency” pattern. Over the thousands of SNP sequences evaluated (which have a near random distribution of each of the homozygote patterns and the heterozygote pattern), the plotted allele peaks pattern that emerges from a balanced (two copies) complement is three distinct lines. If sequences are deleted (only one copy present), the allele peaks pattern that emerges for that region is reduced to two lines (Fig. 7.2). Alternatively, if a trisomic imbalance is present, the allele peaks pattern would demonstrate a gain (four lines) (Fig. 7.3).
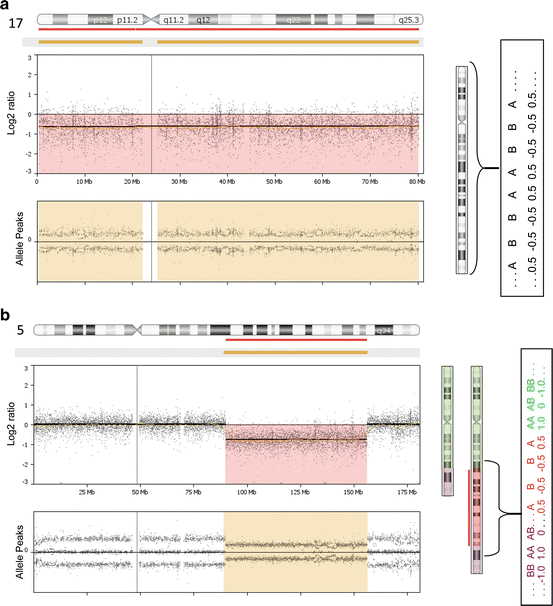
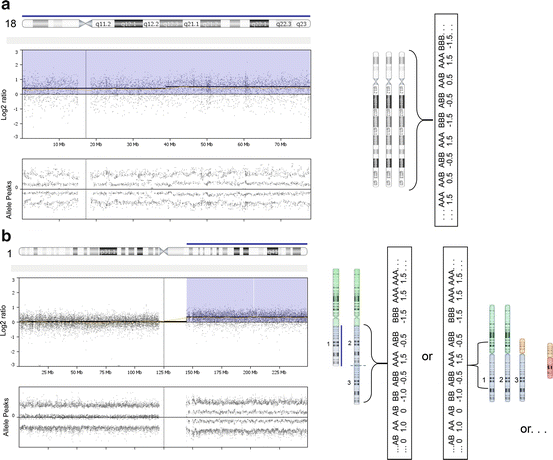
Fig. 7.2
Examples of data patterns observed in solid tumor specimens having monosomic/partial monosomic (one copy) imbalances. The specimens shown in panels (a) and (b) were processed using the Affymetrix OncoScan platform (with the analysis being completed using Nexus Express for OncoScan 3 software). On the left side of the figures, the microarray data is shown for a single chromosome (which is indicated by an ideogram in a horizontal orientation at the top of the figure). (a) Monosomy for chromosome 17 was detected in an FFPE specimen obtained from a spindle cell/pleomorphic lipoma. The red line beneath the horizontal ideogram above the microarray data illustrates that a loss was observed that includes the entire length of the chromosome. The middle frame of this figure shows the log 2 ratio value, which reflects the intensity measures for the probes localized to chromosome 17 for the “test” specimen relative to controls. For specimens having monosomy in 100 % of their cells, one would anticipate a log 2 ratio value approximating −1.0. For this specific specimen, which was comprised primarily of tumor cells, but also contained normal cells, the log 2 ratio value approaches −1.0 (highlighted light red). Since only one chromosome is present in the tumor cells from this specimen, only one allele is present for the sequences localized to chromosome 17 (right hand portion of figure). Therefore, the allele peak pattern for this specimen has two lines with values centering on either 0.5 or −0.5 (lower portion of left handFig. 7.2 (continued) figure that is highlighted in tan). The presence of cell admixture (normal cells and tumor cells) is reflected by the “splitting” of the allele pattern. (b) An interstitial deletion of chromosome 5 (shortened ideogram on right portion of figure) was observed in this specimen. The sequences flanking the breakpoints (designated by green [above breakpoint] or purple [below breakpoint] shading) have a normal complement (two copies of sequences localized to these regions). However, only one copy of sequence is present for a portion of chromosome 5 (highlighted in red on the structurally normal ideogram). Thus, the log 2 ratio value (approximating −1.0; middle panel of left hand figure) and the allele peak pattern (two lines; lower panel of left figure; a single allele on the right ideogram) for this portion of the chromosome are consistent with one copy. The remaining portions of the chromosome have patterns consistent with a balanced (n = 2) complement (log 2 ratio value = 0; Allele peak pattern with three lines; two alleles for each locus)
Fig. 7.3
Examples of data patterns observed in solid tumor specimens having trisomic/partial trisomic (three copies) imbalances. The specimen shown in panels (a) and (b) were processed using the Affymetrix OncoScan platform (with the analysis being completed using Nexus Express for OncoScan 3 software). On the left side of the figures, the microarray data is shown for a single chromosome (which is indicated by an ideogram in a horizontal orientation at the top of the figure). (a) Trisomy for chromosome 18 was detected in an FFPE specimen obtained from a spindle cell/pleomorphic lipoma (same specimen noted in Fig. 7.2). The single blue line above the horizontal ideogram illustrates that three copies were present for the entire length of the chromosome. The middle frame of this figure shows the log 2 ratio value, which reflects the intensity measures for the probes localized to chromosome 18 for the “test” specimen relative to controls. For specimens having trisomy in 100 % of their cells, one would anticipate a log 2 ratio value approximating 0.58 (value consistent with a trisomic imbalance). Given that three chromosomes 18 are present in the tumor cells from this specimen (right side of figure), there are four possible combinations of alleles that could be present. Therefore, the allele peak pattern for this specimen has four lines, with values centering on 1.5, 0.5, −0.5, and −1.5 (lower portion of left hand figure). (b) The specimen evaluated in this frame was a breast cancer tumor. A trisomic imbalance for the long arm of chromosome 1 was observed in this specimen. The sequences localized to the short arm in the ideograms on the right (designated by green shading) have two copies of sequence (log 2 ratio value approximating 0; three lines in the allele peak pattern), with three copies being present for sequences localized to the long arm (highlighted in blue on the ideograms; log 2 ratio value approximating 0.58; four lines in the allele peak pattern). The repeat sequences localized to the pericentromeric region of chromosome 1 are not included in the array (see gap on lower portion of figure) because these sequences are not chromosome-specific (could cross-hybridize to other chromosomes). The microarray does not provide information about the specific type of structural chromosomal abnormality present in this sample. Possible rearrangements include (but are not limited to) a duplication of chromosome 1 (first partial ideogram) or an unbalanced whole arm translocation between chromosomes 1 and 16. In this specimen, a whole arm imbalance was also observed for chromosome 16 (three copies for the short arm [orange] and one copy for the long arm [red]), which suggests that this specimen may have an unbalanced whole arm translocation between chromosomes 1 and 16, the latter of which is a finding observed in breast cancer tumors
Loss of Heterozygosity (or Uniparental Disomy)
In addition to providing information about copy number changes, the microarray platforms that include SNP markers (or probes) allow one to detect loss of heterozygosity (LOH), thereby providing information about a tumor that would not be available from conventional cytogenetic or FISH studies. LOH is detected when a heterozygote has loss of one allele form at a genetic locus. Several different mechanisms can result in LOH, including (but not limited to) chromosome loss following a mitotic nondisjunction or chromosome laggard event, a chromosomal deletion, somatic cell recombination (can be copy neutral), acquired uniparental disomy (copy neutral), or gene conversion (can be copy neutral) [11]. LOH can be seen in many types of tumors and can be associated with a loss of function of tumor suppressor genes. Microarray studies to assess LOH are usually informative directly from the study of the tumor tissue, but may require an additional assessment of normal tissue from the patient to unequivocally categorize the region as having a true loss of heterozygosity versus constitutional absence of heterozygosity [12].
Based on the copy number and SNP patterns present in a tumor, a cytogeneticist is often able to infer the chromosomal alteration(s) leading to the observed imbalance. For example, an imbalance resulting in the loss of one arm of a chromosome, coupled with a gain of the other arm of the same chromosome, is consistent with the presence of an isochromosome. Amplification, which can arise from double minutes, homogeneously staining regions, or complex rings, is often recognized by the presence of heterogeneous patterns (due to the varying number of double minutes, or double rings, ring breakage, etc.) showing a high level of gain (6 or more signals, with some tumors having more than 20 copies of a genomic region) (Fig. 7.4). Microarray studies have also allowed for the recognition of new cytogenetic mechanisms associated with cancer specimens (in both solid tumors and hematologic specimens). One such example is a condition called chromothripsis. The name for this phenomenon was derived from the Greek words “chromos” (which means colored body and reflects the observation that chromosomes stain darkly with certain dyes) and “thripsis” (which means shattering into pieces) [13]. Chromothripsis is visualized by the presence of multiple rearrangements involving either a single chromosome or more than one chromosome. These rearrangements are thought to be initiated by an event that leads to fragmentation of multiple regions of the chromosome(s), with the pieces being reassembled (and/or replicated) erroneously. As a result of the multiple errors in the assembly of the shattered chromosomes, a single chromothripsis event can lead to the loss of multiple tumor suppressor genes, as well as the amplification of multiple oncogenes. Thus, this event can quickly result in aberrant cell growth and can be an early initiating step in cancer [13, 14].
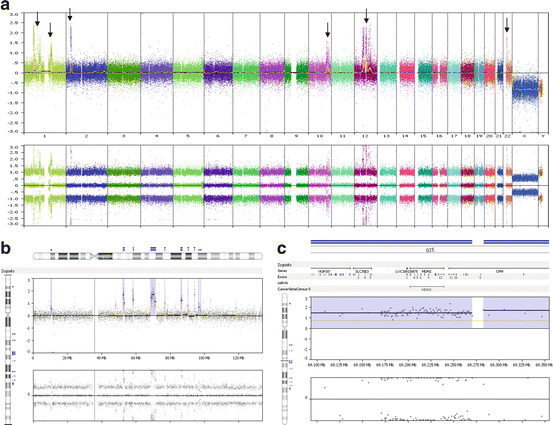
Fig. 7.4
Genome-wide and chromosome-specific results from a microarray study of a dedifferentiated liposarcoma (DNA from FFPE specimen). (a) The genome-wide overview shows multiple areas of imbalance with amplification (highlighted by arrows) including regions localized to chromosomes 1 (includes MDM4), 2 (includes ALK), 10, 12 (includes MDM2), and 22. (b) An assessment of the patterns observed on chromosome 12 shows multiple regions of amplification (areas highlighted by double blue lines) that are present on the long arm (eight regions). (c) The software programs allow one to “magnify” the views to identify genes that are localized to the region(s) of imbalance. In this example, one of the amplicons localized to 12q15 encompasses the MDM2 gene (as well as other genes)
Clearly, the primary clinical goal of DNA copy number and SNP microarray studies of tissue(s) from a solid tumor(s) is to identify genomic imbalances or regions with LOH. While many of the acquired abnormalities identified have known significance to cancer, some may have uncertain clinical relevance. One approach that could provide insight as to the acquired (cancer related) versus constitutional nature of an uncertain copy number alteration is to compare the copy number patterns between paired neoplastic and normal tissue from a patient [109]. However, given that these paired tissue comparisons are quite costly, they are typically done only for the subset of cases lacking clarity (not routinely completed for all specimens) [12]. As our genomic databases expand (they are continuously updated to reflect the discovery of new findings or reclassification of variants), the categorization of variants having clearly pathogenic/benign significance will improve.
Microarray studies (especially the microarrays using SNPs and copy number alterations) also have the potential to provide incidental constitutional information about a patient, including (but not limited to) a history of consanguinity (the patient’s parents may have been related), the presence of constitutional uniparental disomy, a copy number change that might be associated with a late onset health condition, or the presence of a heritable mutation that might predispose the individual [and/or a family member(s)] to develop cancer [15]. It is important that patients with relevant incidental constitutional findings be referred for genetic counseling.
Nomenclature for Microarray Test Results
The nomenclature used to describe the copy number and LOH findings detected in a DNA microarray study has been defined by the members of the International Committee on Human Cytogenetic Nomenclature, with the most recent revision being published in 2013 [110]. This nomenclature system provides a protocol for describing the observed findings using either: (1) a short form (which defines the regions demonstrating the copy number [or absence of heterozygosity] alteration); or (2) a detailed description (which denotes the area showing a change, along with a delineation of the closest bordering nucleotide having a normal result). Using the short form, which is the format used most often by laboratory directors, the nomenclature to designate a normal female complement is:
arr(1–22,X)×2
where “arr” indicates that microarray technology was used; the values in the parenthesis indicate the chromosomes for which data was collected (in this case, chromosomes 1 through 22 and the X chromosome); and the “×2” [e.g., “times 2”] indicates that two copies were observed for all the chromosomal regions interrogated. A normal male complement would be indicated as:
arr(1–22)×2,(XY)×1
where “arr” indicates that microarray technology was used; the chromosomes in the first set of parentheses (chromosomes 1 through 22) were seen in two copies (“×2”), the comma indicates an additional point of information follows, which is shown in the second set of parentheses, the latter of which designates that each sex chromosome (X and Y within the parenthesis) is present in 1 copy (“×1” means times one copy). If a copy number change is observed, the variant is designated by listing the chromosome or chromosomal band/bands. If the copy number alteration involves the entire chromosome, the nucleotides are not listed. For example, for copy number changes resulting in a monosomic or trisomic complement, the chromosome having the aneuploid complement would be noted in parenthesis, followed by the number of copies for that chromosome:
Copy number alterations often involve only a portion of the chromosome. The nomenclature to describe a structural rearrangement includes a designation of the chromosomal band(s) having an imbalance, followed by a listing of the nucleotides encompassed in the imbalance (map coordinates), and the number of copies present for the designated region, as shown below:
arr 5q14.3q33.3(89,979,589–156,261,737)×1 (Fig. 7.2)
which indicates that a microarray study was completed (arr) that showed an imbalance involving bands 5q14.3 to 5q33.3 (where “q” is the abbreviation for the long arm); this imbalance encompassed 66,282 kb (156,261,737 minus 89,979,589; divided by 1,000 since the number of nucleotides is described in kilobases in this example) and showed a copy number loss (one copy, as indicated by “×1”). A copy number variation involving gain of a portion of a chromosome is indicated using the same logic as that used for loss, as shown in the example below:
arr 1q21.1q44(145,115,883–249,212,878)×3 (Fig. 7.3)
which indicates that a microarray study was completed (arr) that showed an imbalance involving bands 1q21.1 through 1q44; this imbalance encompassed 104,097 kb (249,212,878 minus 145,115,883; divided by 1,000 since the number of nucleotides is described in kb in this example) and showed a trisomic imbalance (three copies as indicated by “×3”).
In addition to providing a means to designate copy number alterations, the nomenclature also provides a means for describing a long contiguous stretch (or stretches) of homozygosity. Several terms can be used to describe this finding, including (but not limited to) LOH [term often used in cancer] or absence of heterozygosity [term sometimes used when long contiguous stretches of homozygosity are observed as a constitutional finding]. Solid tumors often have complex cytogenetic findings, with more than one imbalance per chromosome. In these cases, the nomenclature will reflect each of the aberrations present, as shown in the example below denoting three findings localized to a chromosome 5 (Figs. 7.5 and 7.6):
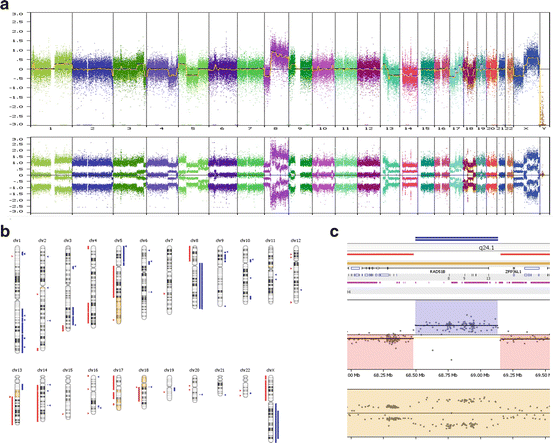
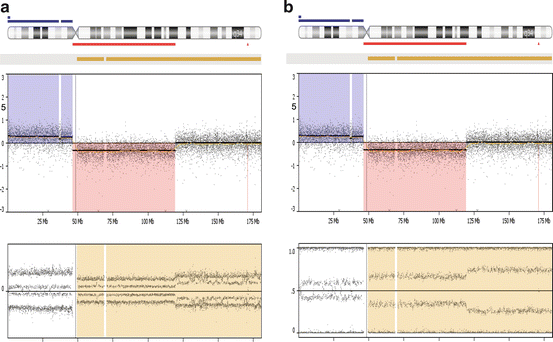
Fig. 7.5
Genome-wide and chromosome-specific results from a microarray study of a breast cancer tumor (DNA from FFPE specimen with 55 % of cells being derived from tumor). (a) The log 2 ratio and allele peak values are shown for each of the chromosomes (differentially colored). This specimen has a complex karyotype with multiple aberrations involving each of the 22 autosomes and the X chromosome, with these anomalies including regions of amplification, trisomy, loss, or LOH. (b) The findings present in this same tumor are presented in a format that summarizes the regions showing gains (blue), loss (red), and copy neutral LOH (yellow). (c) A focal region of high gain (double blue line with five copies that also shows an LOH) encompassing the RAD51 gene (which functions in DNA repair) was observed at band 14q24.1, with this area being juxtaposed to regions having loss
Fig. 7.6
Results from a microarray study showing loss, gain, and LOH involving different regions from chromosome 5. The DNA used for the microarray study of this breast cancer tumor (same specimen as shown in Fig. 7.5) was extracted from FFPE tissue that had 55 % tumor cells present. The abnormalities observed for chromosome 5 from this specimen included regions showing gain that involved the short arm, regions showing loss that involved the proximal to mid long arm, and regions with a copy neutral loss of heterozygosity. This same data is shown as it appears using either an allele peak format (a) or a B-allele frequency format (b). For both presentations, the values lie mid-way between the anticipated values for trisomy, monosomy, and LOH due to the admixture of tumor (estimated to be present in 55 % of cells) and normal cells that were present in this heterogeneous sample following macrodissection, resulting in a “mosaic” allele band pattern due to the presence of both normal and abnormal cells
arr 5p15.33p13.2(1,854,650–36,656,718)×2 ~ 3,5p11q23.1(46,401,271–119,640,226)×1 ~ 2, 5q23.1q35.3(119,640,227-180,698,312)×2 hmz
which indicates that a microarray study was completed (arr) that showed a 34,802 kb region (36,656,718 minus 1,854,650; divided by 1,000 since the number of nucleotides is described in kilobases in this example) with three copies of the sequences localized to bands 5p15.33 to 5p13.2 being present in a portion of cells (in this case, three copies were present in 55 % of the cells, with 45 % of cells having two copies due to the specimen containing both tumor and normal cells, but the percentage of cells is not currently designated in the nomenclature). A second imbalance involving a deletion (1 copy) that encompassed 73,239 kb (119,640,226 minus 46,401,271; divided by 1,000 since the number of nucleotides is described in kb in this example) of sequences localized to bands 5p11 to 5q23.1 was also seen in a portion of cells (as noted above, in this specimen 55 % of the cells had the abnormality, with one copy likely present in the tumor cells). A third finding involving chromosome 5 was a 61,058 kb long contiguous stretch of homozygosity (hmz), which is also referred to as LOH, that was localized to bands 5q23.1 through 5q35.3. This LOH is described as being copy neutral since there are two copies of these sequences. In addition to having multiple aberrations involving a single chromosome, one can also have multiple chromosomes that demonstrate findings. In specimens having multiple rearrangements throughout the genome, the nomenclature can be indicated as:
arr(1–22,X)cx (Fig. 7.5)
where “cx” stands for complex. In this example the specimen was collected from a female and the aberrations involved the autosomes and the X chromosomes. A similar complex complement observed in a specimen collected from a male would be designated as “arr(1–22,XY)cx”. For complex tumors, the cytogeneticist/geneticist evaluating the specimen may elect to list the individual findings or describe them in a tabular format to facilitate their recognition by health care providers managing the patient’s care.
Along with using the nomenclature (and/or a supplemental format) to note the microarray test results, most reports will also include an interpretative section to describe the findings and to relate the observations to their clinical significance. In addition, the report may provide a recommendation for reflex testing that might be helpful for optimal interpretation of the results, such as FISH studies to confirm the microarray results and/or establish the pattern present in the tumor. Also, some reports may include information about prognostic indications or therapeutic regimens that have been associated with the observed findings to aid the clinician in their provision of care. A resource that may be of value to clinicians (and laboratory directors/pathologists) in identifying targeted therapies related to the observed genomic imbalances was developed by scholars at Vanderbilt University and is freely available at http://bioinfo.mc.vanderbilt.edu/software.html.
Specimen Selection
As noted above, a key component to the success of the microarray methodology is the use of high quality DNA that is not degraded. When studying solid tumors, the most frequently utilized specimen sources include (but are not limited to) DNA obtained directly from fresh tumor tissue, DNA obtained from a rapidly frozen specimen (also called snap frozen), or DNA obtained from a formalin-fixed, paraffin-embedded specimen (FFPE). Advantages of using an FFPE specimen include: (1) the tissue does not require any additional processing steps beyond those typically followed in a pathology lab; (2) one has the ability to identify the location of the tumor in an adjacent H&E stained slide and macro-dissect the specimen to enrich for the presence of tumor cells in the tissue used for DNA extraction; and (3) one can characterize and compare copy number and LOH alterations present in tissues having differing histological morphologies [16]. Furthermore, use of FFPE cases provides an opportunity to determine DNA copy number alterations/LOH for archival specimens, thereby providing a means to obtain long-term clinical follow-up information and/or to amass a larger number of rare tumors for study.
While the FFPE specimens have some clinical advantages when compared to the fresh or frozen tissue, they also present technological challenges since the processing for FFPE tissue leads to degradation of its DNA. Indeed, investigators have consistently shown that fresh or rapidly frozen tissue tends to yield better quality DNA. One approach that has been developed to circumvent the technical hurdle of DNA degradation in FFPE tissue is the use of inversion probes [9]. An alternative strategy that has been used involves “repairing” degraded DNA [17]. When determining the protocol a lab elects to implement for solid tumor tissue collection, one should consider the quality of the test result as well as the ease and timeliness of obtaining tissue. Based on the workflow of the center, the microarray testing lab should then validate their selected solid tumor microarray platform (studying at least 30 tumors) following the clinical practice and tissue acquisition protocol deemed optimal for their provision of patient care at their hospital and/or referral sites.
Literature Review of Microarray Studies of Solid Tumors
Copy Number Changes
DNA copy number microarray studies of solid tumors have not only resulted in refinement of the breakpoints of genes from previously known pathognomonic alterations, but have also allowed for the identification of heretofore unrecognized abnormalities associated with specific types of tumors. In their eloquent study integrating data from The Cancer Genome Atlas (TCGA), Ciriello et al. [18] identified emerging patterns of genomic alterations from their hierarchical classification of more than 3,000 tumors that were obtained from 12 types of cancer, including 11 types of solid tumors. They determined that tumors could be classified into two broad categories, with this distinction being driven by the presence/absence of a TP53 mutation(s). The tumors lacking a TP53 mutation showed primarily somatic mutations (“M” class). In contrast, the tumors that were positive for a TP53 mutation contained primarily chromosomal imbalances (“C” class). Among the C class tumors, they observed that cancer specimens originating from different tissues often shared similar chromosomal alterations; yet, paradoxically, different tumors from the same tissue could show significant variation. The DNA imbalances that were observed most frequently included those involving losses of chromosome bands 9p21 (including the CDKN2A gene), 8p23, and 8p21. Gains of chromatin were observed most often for chromosome bands 3q26, 8q24 (MYC gene), 1q21, 11q13 (CCND1 gene), 8q22, 5p15, 7p11 (EGFR gene), 20q13, 19q12 (CCNE1 gene), 8p11, 1q21, and 6p23 (E2F3 gene) [18].
Our review of the extant literature of copy number changes identified in solid tumors following microarray evaluations echoed the observations of the Ciriello team and resulted in the recognition of recurrent “hot spots” for chromosomal imbalances. Tumors from patients diagnosed with colorectal cancer are among those most frequently evaluated using DNA copy number microarray technology (Table 7.1). The consistent regions with chromosomal imbalances that have been recognized from these studies include: (1) gains involving 7p, 8q, 13q, and/or 20q; and (2) losses of 1p, 4 (p and q), 8p, 14q, 17p, and/or 18 (p and q). Studies using conventional cytogenetic and FISH methodologies have also shown a nonrandom association of these chromosomal imbalances in colorectal cancer tumors. In addition to these consistent observations, studies of DNA copy number microarrays have provided new insight regarding the increased frequency of cytogenetic aberrations in tumors lacking microsatellite instability when compared to tumors with microsatellite instability [19, 20] (Table 7.1). Microarray studies have also resulted in the discovery of a nonrandom association of a deletion of the WWOX tumor suppressor gene (localized to 16q23.1) in colorectal cancer tumors having microsatellite instability [19]. Studies of copy number DNA alterations have also contributed to advances in prognostication for patients with colorectal cancer. For example, patients having simultaneous deletions involving 18q, 8p, 4p, and 15q have been recognized as a subgroup having a poorer prognosis when compared to other patients [21, 22].
Table 7.1
Copy number alterations detected in colorectal tumors/cell lines using DNA microarray methodology
Author (year) | PMID # | Specimen type | Platform | Copy number alterations | |
|---|---|---|---|---|---|
Gains | Losses | ||||
Gaasenbeek et al. (2006) [41] | 16585170 | 45 cell lines | Affymetrix SNP GeneChip Human 10 K | – | 17p, 18q |
Tsafrir et al. (2006) [42] | 16489013 | 7 primary colon tumorsa, b | Affymetrix GeneChip Human Mapping 50 K | 7p, 8q,13q, 20q | 1p, 4, 5q, 8p, 14q, 15q, 18 |
Andersen et al. (2007)[43] | 16774939 | 15 FFc | Affymetrix Genechip Mapping 10 K | 7p, 8q, 13q, 20q | 1p, 4, 5q, 8p, 10 14q, 15q, 17p, 18, 21q |
Lips et al. (2007) [44] | 17471469 | 78 FF rectal adenomas and carcinomas | Affymetrix GeneChip Mapping 10 K | 8q, 13q, 17p, 18q, 20q | 1p, 4q, 5q, 7p, 12q |
Kurashina et al. (2008)[45] | 18564138 | 92 FF sporadic cases | Affymetrix 50 K GeneChip | 6, 7p, 8q, 20q | 1p, 3, 5q, 8p, 14q, 17p, 18, 20 |
Sheffer et al. (2009) [21] | 19359472 | 62 primary carcinomasa; 8 liver metsa, d; 10 lung metsa | Affymetrix 50 K GeneChip | 7, 8q, 13q, 20, X | 4, 8p, 14q, 15q, 17p, 18, 20p, 22q |
Sayagués et al. (2010)[46] | 21060790 | 23 FF primary tumors | 250 K Affymetrix SNP Mapping | 1q, 7, 8q, 11q, 13q, 20q, X | 1p, 8p, 14q, 17p, 18q, 22q |
Lin et al. (2011) [20] | 21645411 | 16 MSIe 13 MSSe | Affymetrix Human SNP 6 | MSS: 7, 8q, 13, 20 | MSS: 8p, 18 |
Xie et al. (2012) [19] | 22860045 | 302 FFPEf | Affymetrix OncoScan V1.0 | MSS –7p, 8q, 13q, 20q | MSI: 8p, 17p, 18q, 21q MSI – 16q, 20q |
Jasmine et al. (2012) [47] | 22363777 | 86 FF | Illumina SNP | 5p, 8q, 13q, 20q | 5p, 8p, 17p, 18 |
Middeldorp et al. (2012)[48] | 21445971 | 30 FFPE from familial CRCs | Illumina 6 K | 7, 8q, 13q, 20 | 17p, 18 |
Chen et al. (2013) [49] | 23434627 | 126 FF | CytoScan HD | 7p, 8q, 13q, 20q | 8p, 15q, 17p, 18q |
Eldai et al. (2013) [50] | 24204606 | 15 primary FF | CytoScan HD | 7, 8q, 12, 13, 20q | 1, 4, 6, 10, 14q, 17p, 18, 21 |
For breast cancer, the imbalances observed following DNA copy number microarray studies are largely consistent with the imbalances that have been most frequently observed in these tumors using GTG-banding and/or FISH methodologies. Specifically, recurrent imbalances detected with DNA copy number microarray methodology in breast cancer tumors include gains for regions localized to 1q, 2p, 8q, and/or 17q (including, but not limited to, ERBB2 (HER2) amplification) as well as losses involving regions localized to 1p, 2p, 9p, and/or 16q (Table 7.2). The gain of 1q and loss of 16q is consistent with the observation of an unbalanced whole arm translocation between the long arm of chromosome 1 and the short arm of chromosome 16, with this unbalanced rearrangement being one of the most frequently noted cytogenetic anomalies observed in breast carcinomas [23]. Microarray studies have also allowed for clarification of the size of the region amplified in ERBB2 positive tumors. The amplicons in ERBB2+ tumors have been noted to range in size from less than 86 kb to several hundred kb and to encompass not only the ERBB2 (HER2) gene, but also several other genes (including, but not limited to, the TCAP, PNMT, PGAP3 (PERLD1), GRB7, RARA, and TOP2A genes) [24, 25]. Microarray studies have also contributed to improvements in prognostication for patients with breast cancer, as well as insight regarding the types of genomic imbalances present in subclasses of tumors [26–31]. For example, women with breast tumors having amplification for the MET and/or PIK3CA gene have been noted to have a poorer prognosis, with these amplifications tending to be seen more often in triple negative tumors [30]. Moreover, when PIK3CA amplification is acquired in ERBB2+ tumors, these patients show disease progression, with associations between PIK3CA amplification and treatment resistance being reported [31]. DNA copy number microarray methods have also been used to study the constitutional genomic makeup of people with a family history of breast cancer, with the goal of identifying potential copy number variants that might confer a predisposition (or reduction) for one’s risk to develop breast cancer [32, 33].
Table 7.2
Copy number alterations detected in breast tumor tissues/cell lines using copy number microarray methodology
Author (year) | PMID # | Specimen type | Platform | Copy number changesa | |
|---|---|---|---|---|---|
Gains | Losses | ||||
Bergamaschi et al. (2006)[26] | 16897746 | 89 FFb | Custom Array (22,388 sites) | 1q, 8q, 11q, 16p | 4q, 5q, 6q, 8p, 14q |
Riener et al. (2008) [51] | 18656243 | 21 FFPEc (Tubular) | GenoSensor Array 300 microarray | 1p/1q, 2p, 2q, 5p, 6p/q, 7q, 8p, 8q, 11p, 13q, 15q, 17q, 18q, 20p, 22q | 1p, 2p, 2q, 4q, 6p, 7q, 9p, 10q, 12q, 15q, 16q, 19q, 20q, Xq |
Argos et al. (2008) [52] | 18406867 | 16 FF | Affymetrix 10 K SNP array | 1q | 2p, 2q, 16q |
Staaf et al. (2010) [24] | 20459607 | 200 FF | Custom BAC array for chromosome 17 | Higher copy numbers (amplification) of HER2+ associated with poorer prognosisd | – |
Brewster et al. (2011)[53] | 21795423 | 850 FFPE | Affymetrix OncoScan | 2p, 3q, 8q, 11p, 20q seen more often in symptom-detected (versus screen detected) tumors | – |
Johnson et al. (2012) [54] | 22052326 | 21 FFPE synchronous ductal carcinoma in situ (DCIS) and invasive ductal carcinoma (IDC) specimens | Affymetrix OncoScan | DCIS: No type-specific gains noted IDC: 5q, 16p, 19q, 20 | DCIS: 17p IDC: No type-specific losses noted |
Thompson et al. (2011) [29] | 21858162 | 971 FFPE | Affymetrix OncoScan | Xq, 1q, 2p, 3q, 4q, 5p, 8q, 10p, 10q, 11q, 12p, 14q, 17q, 20qe | 1p, 6q, 8p, 9p, 11p, 12q, 13q, 16p, 22qe |
In addition to studies of more frequent tumors, microarray studies have been completed for a variety of solid tumors, including rare tumors (Table 7.3). Microarray technology is especially helpful for gaining knowledge about chromosomal imbalances present in cells from tumors that have been difficult to successfully evaluate using in vitro cell culturing/chromosomal methods. In one of the most comprehensive reports of genomic imbalances observed in different types of tumors using DNA copy number and SNP microarray methodology, Dougherty et al. [12] reported findings for a total of 168 pediatric tumors and found abnormalities in 82 % of the specimens. These investigators also underscored the contribution of DNA copy number arrays for the study of solid tumors, noting that abnormal microarray results were successfully obtained for 63 specimens (37 % of the total sample) that could not be analyzed (due to growth compromises and other technical challenges) using conventional cell culture/chromosomal banding methodology. Furthermore, they showed that the improved resolution and genome-wide coverage of microarrays allowed for the detection of aberrations that were not seen in the chromosomal or FISH studies.
Table 7.3
Copy number alterations detected in studies of various solid tumors evaluated using genome-wide copy number microarrays
Author (year) | PMID # | Specimen type | Platform | Copy number changes | |
|---|---|---|---|---|---|
Gains | Losses | ||||
Adenoid cystic carcinoma | |||||
Yu et al. (2007) [55] | 17372589 | 22 primary FFa; 2 cell lines | Affymetrix GeneChips Human Mapping 100 K | ACC3 cell line: 2p, 7p | 9p |
Adrenocortical | |||||
Stephan et al. (2008) [56] | 18281524 | 21 FF; 4 (not specified) | Agilent Human Genome Microarray Kit 44B | 5, 6q, 7, 8q, 12, 16q, 20 | 1, 2q, 3, 6p, 7p, 8p, 9, 10, 11, 13q, 14q, 15q, 16, 17, 19q, 22q |
Astrocytoma | |||||
Sievert et al. (2009) [57] | 19016743 | 28 FF | Illumina HumanHap550K SNP | 7q34 | |
Schiffman et al. (2010) [58]
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |||||