Antiphospholipid syndrome (APS) is a systemic disease that causes venous and arterial thrombosis in virtually any organ and is responsible for fetal losses and pregnancy disorders. Previously, APS was thought to be present mainly in patients with systemic lupus erythematosus. The spectrum of clinical manifestations is wide, because the thrombotic process may involve arterial and venous vessels of any size in any organ. At present, there is no evidence to support or refute specific treatment strategies for primary prophylaxis of thrombosis.
Antiphospholipid syndrome (APS) is a systemic disease that causes venous and arterial thrombosis in virtually any organ and is responsible for fetal losses and pregnancy disorders. The recognition of APS as an independent nosologic entity is relatively recent, dating back to the late 1980s. Previously, APS was thought to be present mainly in patients with systemic lupus erythematosus (SLE). During the 1970s and 1980s, there were already case reports or small case series describing the association between thrombotic events, fetal loss, and a circulating lupus anticoagulant (LA), without SLE. However, it was only in the late 1980s that the existence of a primary APS (ie, in absence of SLE) was postulated. An international consensus statement on classification criteria of APS was published in 1999 and updated in 2006. Antiphospholipid antibodies (aPL-Abs) constitute a heterogeneous family of autoantibodies directed against proteins that bind to anionic phospholipids and are the basis of the prothrombotic state that characterizes APS.
The spectrum of clinical manifestations is wide, because the thrombotic process may involve arterial and venous vessels of any size in any organ; deep vein thrombosis (DVT) of the lower limbs, pulmonary embolism (PE), stroke or transient ischemic attack (TIA), and myocardial infarction (MI) are some of the most common thrombotic events. Fetal manifestations include early and late fetal loss and premature birth, representing a frequent manifestation of APS in women of childbearing age.
Despite important research efforts over the last 15 years, many questions remain unaddressed, regarding the important aspects of pathophysiology, treatment, and prognosis of this peculiar type of acquired thrombophilia.
Epidemiology
Data on the prevalence of aPL-Abs should be considered with caution, giving the incomplete standardization of methods for their detection. The prevalence of aPL-Abs ranges from 1% to 10% in general population (with an even higher proportion among the elderly) and from about 30% to 40% (and even higher) in patients with SLE. The risk of thrombotic events in asymptomatic patients with aPL-Abs can be up to about 3% per year. Patients presenting with thrombosis have a prevalence of up to 30%. In women with recurrent fetal losses, the proportion of aPL-Abs has been described to vary between 5% and 60%, reflecting again a lack of standardization in aPL-Abs detection (about 15% had persistent aPL-Abs). In unselected women, aPL-Abs were found in 3% to 5%; screening-detected aPL-Abs did not predict poor pregnancy outcome.
Pathophysiology
aPL-Abs are autoantibodies directed against proteins that bind to anionic phospholipids. Besides APS, aPL-Abs are also found in patients with other conditions including cancer, infections, and in concert with the use of selected medications.
Many antigens have been described to be recognized by aPL-Abs, including β 2 glycoprotein I (β 2 GPI), prothrombin (PT), protein C complex, annexin A5, proteins of the coagulation cascade (factors VII, XI, XII), and proteins of the fibrinolytic system.
The exact mechanisms by which aPL-Abs cause thrombosis and other clinical manifestations of APS are not understood. The effects of aPL-Abs are directed at different pathways involved in the coagulation process: inhibition of natural anticoagulants and the fibrinolytic system and activation of endothelial cells, platelets, and the complement system.
β 2 GPI, a plasma protein with affinity for anionic membrane phospholipids, binds to such membranes with high affinity when aPL-Abs and anti-β 2 GPI antibodies are present, affecting the coagulation or fibrinolysis process on those cellular surfaces. Effects may include interference with protein C and other coagulation proteins. Some anticardiolipin antibodies (aCL-Abs) are directed against PT; they may elicit a prothrombotic effect by promoting the activity of PT and interfering with the action of activated protein C (which is a normal indirect inhibitor of thrombin).
aPL-Abs can also react with plasmin, downregulating clot lysis. They may also inhibit the activity of tissue factor pathway inhibitor, an early inhibitor of coagulation activation.
aPL-Abs can also activate endothelial cells, which in turn increase the expression of cellular adhesion molecules and tissue factor, thus enhancing the activation of the coagulation process. Finally, the effects of aPL-Abs on platelets include an increase of GpIIb-IIIa expression and increase of thromboxane A 2 synthesis, stimulating platelet aggregation.
The exact mechanism of fetal loss remains still largely unknown. One possible mechanism involves β 2 GPI that binds to anionic phospholipids during trophoblast differentiation. When aPL-Abs are present, they can bind to β 2 GPI on the membrane of trophoblast cells, thus interfering with adequate placentation. Other possible mechanisms, studied in animal models, involve complement activation (causing fetal damage) and inhibition of annexin A5 (a placental anticoagulant protein) by aPL-Abs.
Pathophysiology
aPL-Abs are autoantibodies directed against proteins that bind to anionic phospholipids. Besides APS, aPL-Abs are also found in patients with other conditions including cancer, infections, and in concert with the use of selected medications.
Many antigens have been described to be recognized by aPL-Abs, including β 2 glycoprotein I (β 2 GPI), prothrombin (PT), protein C complex, annexin A5, proteins of the coagulation cascade (factors VII, XI, XII), and proteins of the fibrinolytic system.
The exact mechanisms by which aPL-Abs cause thrombosis and other clinical manifestations of APS are not understood. The effects of aPL-Abs are directed at different pathways involved in the coagulation process: inhibition of natural anticoagulants and the fibrinolytic system and activation of endothelial cells, platelets, and the complement system.
β 2 GPI, a plasma protein with affinity for anionic membrane phospholipids, binds to such membranes with high affinity when aPL-Abs and anti-β 2 GPI antibodies are present, affecting the coagulation or fibrinolysis process on those cellular surfaces. Effects may include interference with protein C and other coagulation proteins. Some anticardiolipin antibodies (aCL-Abs) are directed against PT; they may elicit a prothrombotic effect by promoting the activity of PT and interfering with the action of activated protein C (which is a normal indirect inhibitor of thrombin).
aPL-Abs can also react with plasmin, downregulating clot lysis. They may also inhibit the activity of tissue factor pathway inhibitor, an early inhibitor of coagulation activation.
aPL-Abs can also activate endothelial cells, which in turn increase the expression of cellular adhesion molecules and tissue factor, thus enhancing the activation of the coagulation process. Finally, the effects of aPL-Abs on platelets include an increase of GpIIb-IIIa expression and increase of thromboxane A 2 synthesis, stimulating platelet aggregation.
The exact mechanism of fetal loss remains still largely unknown. One possible mechanism involves β 2 GPI that binds to anionic phospholipids during trophoblast differentiation. When aPL-Abs are present, they can bind to β 2 GPI on the membrane of trophoblast cells, thus interfering with adequate placentation. Other possible mechanisms, studied in animal models, involve complement activation (causing fetal damage) and inhibition of annexin A5 (a placental anticoagulant protein) by aPL-Abs.
Clinical manifestations
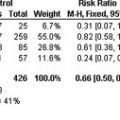
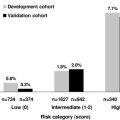
APS has many different clinical manifestations, with a tendency to recur after the initial APS diagnosis. Thrombotic events, involving arteries, veins, or small vessels in any organ or tissue, are the basis of clinical manifestions of APS. In a large, multicenter, international cohort study enrolling 1000 patients with a mean age of 34 years at APS onset, the most frequent thrombotic events at presentation were DVT (31.7%), stroke (13.1%), superficial vein thrombosis (9.1%), PE (9%), TIA (7%), and MI (2.8%).
Other frequent manifestations at onset were thrombocytopenia (21.9%) and livedo reticularis (20.4%); less frequent manifestations were hemolytic anemia, skin ulcers, and pseudovasculitic skin lesions, amaurosis fugax, digital gangrene, and epilepsy. Of note, in this cohort of patients with APS, coexistence of SLE or lupuslike syndrome or other rheumatologic disorders was documented in 47% of patients.
With regard to obstetric manifestations, fetal loss was present in 10% of women as the first clinical manifestation. From APS onset until entry into the study (mean period of evolution, 92 months), 591 women (71.9% of total number of women) experienced at least 1 pregnancy, with 1580 pregnancies in total. Among them, there were 560 (35.4%) early fetal losses (<10 weeks of gestation), 267 (16.9%) late fetal losses (≥10 weeks), and 753 (47.7%) live births (10% of which were premature).
When considering the cumulative clinical features of patients in that cohort, from disease onset until entry into study (mean period of evolution 92 months), venous thromboembolism (VTE), stroke or TIA, MI, fetal losses, livedo reticularis, and thrombocytopenia were still the main clinical manifestations. Of interest, there was a long series of other less-frequent thrombotic events, for example, arterial thrombosis of upper and lower limbs, cerebral venous thrombosis, renal thrombotic manifestations involving renal vein or artery or glomerular vessels, mesenteric ischemia, Budd-Chiari syndrome, and retinal artery and vein thrombosis.
Other less common clinical features deserve some attention, because they were present during the period of evolution in a substantial proportion of patients: migraine (20.2%), epilepsy (7%), cardiac valve thickening or dysfunction (11.6%), amaurosis fugax (5.5%), and hemolytic anemia (9.7%).
APS can also present, at onset or some time after diagnosis, with catastrophic APS (CAPS), which accounts for about 1% of APS cases. Patients with CAPS present with rapidly progressive multiorgan involvement with histopathologic evidence of small vessel occlusions. Despite treatment, CAPS is a life-threatening disease with a mortality of about 50%. Data from an ongoing international registry suggest that 46% of patients develop CAPS de novo, without any previous diagnosis of APS. Intra-abdominal manifestations were present in most patients, involving kidney (71%), liver (33%), gastrointestinal tract (25%), spleen (19%), adrenal gland (13%), and pancreas (8%). Pulmonary involvement (mainly acute respiratory distress syndrome and PE) was present in 64% of patients, cerebral manifestations (mainly multiple microinfarctions as well as seizures and headache) in 62%, cardiac disease (cardiac failure, MI, valve lesions) in 51%, and skin complications (livedo reticularis, skin necrosis) in 50%.
Diagnosis
After a preliminary consensus statement was formulated in Sapporo, Japan in 1998, the diagnostic criteria for APS were revised in Sydney, Australia in 2005. The consensus statement contains clinical criteria for thrombotic events and pregnancy complications and laboratory criteria. The diagnosis of APS is confirmed in the presence of at least 1 clinical and 1 laboratory criterion.
Clinical Criteria
- 1.
Vascular thrombosis: one or more venous, arterial, or small vessel thrombotic events in any tissue or organ (superficial vein thrombosis is not included in clinical criteria)
- 2.
Pregnancy morbidity
One or more unexplained deaths of a morphologically normal fetus at or beyond the 10th week of gestation
One or more premature births of a morphologically normal neonate before the 34th week of gestation because of eclampsia, severe preeclampsia, or placental insufficiency
Three or more consecutive spontaneous abortions before the 10th week of gestation (in the absence of parental chromosomal causes and maternal anatomic or hormonal abnormality).
Laboratory Criteria
- 1.
LA present in plasma on 2 or more occasions at least 12 weeks apart (measured according to the guidelines of the International Society on Thrombosis and Haemostasis )
- 2.
aCL-Abs (IgG and/or IgM) present in serum or plasma on 2 or more occasions at least 12 weeks apart (in medium or high titer, ie, >40 GPL [IgG phospholipid] or MPL [IgM phospholipid] units or >99th percentile, measured by a standardized enzyme-linked immunosorbent assay [ELISA])
- 3.
Anti-β 2 GPI antibody (IgG and/or IgM) present in serum or plasma on 2 or more occasions at least 12 weeks apart (in titer >99th percentile, measured by a standardized ELISA).
The diagnosis of APS cannot be confirmed if less than 12 weeks (or more than 5 years) separate the time of the positive aPL-Ab test and the time of the clinical manifestation.
Moreover, as stated for laboratory criteria, persistence of the aPL-Abs and a second positive test, at least 12 weeks after the first, are required for the diagnosis. To address the issue of epiphenomenal aPL-Abs, the interval required to confirm positivity was increased from 6 weeks (Sapporo criteria) to 12 weeks (revised criteria). However, these intervals are based on expert opinions, confirming the need for validation studies. Coexistence of SLE (or other diseases) distinguishes between primary and secondary APS.
Preliminary diagnostic criteria for CAPS were proposed in 2003:
- 1.
Evidence of involvement of 3 or more organs, systems, and/or tissues
- 2.
Development of manifestations simultaneously or in less than a week
- 3.
Confirmation by histopathology of small vessel occlusion in at least 1 organ or tissue
- 4.
Laboratory confirmation of the presence of aPL-Abs.
The diagnosis of definite CAPS requires all 4 criteria to be fulfilled. In patients without a history of APS, laboratory confirmation of the presence of aPL-Abs must occur and must be confirmed again at least 6 weeks later.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


