SUMMARY
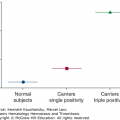
Clinically significant autoantibodies to coagulation factor deficiencies are uncommon but can produce life-threatening bleeding and death. The most commonly targeted coagulation factor in autoimmunity is factor VIII. Acquired hemophilia A, which results from these antibodies, can either be idiopathic or associated with older age, other autoimmune disorders, malignancy, the postpartum period, or the use of drugs such as penicillin and sulfonamides. Bleeding in acquired hemophilia A is treated with factor VIII–bypassing agents. The underlying autoimmune disorder frequently responds to immunosuppressive medication. Antiprothrombin antibodies usually are found in patients with lupus anticoagulant and are associated with bleeding. Antibodies of von Willebrand factor are found in patients with type 3 von Willebrand disease in response to infusion of plasma concentrates containing von Willebrand factor. Antibodies to factor V can occur as autoantibodies or as cross-reacting antibovine factor V antibodies that develop after exposure to bovine thrombin products that are contaminated with factor V. Pathogenic autoantibodies also have been described that target thrombin, factor IX, factor XI, factor XIII, protein C, protein S, and the endothelial cell protein C receptor.
Antibodies directed against coagulation factors can develop as an acquired, autoimmune phenomenon. These “circulating anticoagulants” or “inhibitors” were recognized as early as 1906 as a cause of an acquired bleeding disorder.1 The most common coagulation factor targeted in autoimmunity is factor VIII. The key feature that distinguishes antibody-mediated from other acquired coagulation factor deficiencies, such as impaired synthesis (e.g., a result of vitamin K deficiency) or increased consumption (e.g., in disseminated intravascular coagulation), is the ability of the patient’s plasma to inhibit the coagulation of normal plasma. Inhibitors also can develop in response to replacement therapy in patients with congenital coagulation factor deficiencies, as discussed in Chap. 13.
Acronyms and Abbreviations:
APC, activated protein C; aPCC, activated prothrombin complex concentrate; aPTT, activated partial thromboplastin time; BU, Bethesda units; CTLA4, cytotoxic T-lymphocyte associated protein 4; DAMP, damage-associated molecular patters; EACH, European Acquired Hemophilia Registry; FEIBA, factor VIII inhibitor–bypassing agent; PAPP, pathogen-associated molecular patterns; rVIIa, recombinant activated factor VII.
The incidence of autoantibodies to factor VIII, which is the most commonly targeted coagulation factor in autoimmunity, is 1.4 per million people per year.2–4 The associated clinical condition is called acquired hemophilia A. Approximately 40 to 50 percent of acquired hemophilia A patients have underlying conditions, including other autoimmune disorders (e.g., rheumatoid arthritis, systemic lupus erythematosus), malignancy, pregnancy, or a history consistent with a drug reaction.5 The remaining idiopathic cases most commonly occur in elderly patients of either sex, with the median age at diagnosis being in the mid-70s.
Even though adaptive immunity provides a unique ability to recognize a nearly infinite range of antigenic determinants, mechanisms of immunologic tolerance exist that reduce the probability of autoimmunity.6 Self–nonself discrimination provides the key foundation upon which immune activity can be specifically directed toward potential pathogens.6 However, self–nonself discrimination alone does not possess the capacity to distinguish innocuous antigens from antigens associated with a real threat of infection.7 As a result, an elaborate network of innate immune factors also exists, which recognize potential danger in the form of cellular injury or conserved determinants on pathogens themselves, often referred to as damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), respectively.7,8 Activation of immune cell function following exposure to PAMPs or DAMPs provides the necessary signals required for an efficient immunologic response to foreign antigen.7–9
The development of anti–factor VIII antibodies following factor VIII infusion in individuals with hemophilia A provides a classic example of the deleterious outcome of alloantibody formation following exposure to alloantigen. In this scenario, the factor VIII protein is foreign to the patients; consequently, central tolerance to the factor VIII protein does not occur. In contrast, acquired hemophilia results from loss of previous tolerance to a self antigen.10–12
For alloantibody development in patients with hemophilia A, individual variability in factor VIII levels accounts for some of the divergent level of tolerance to factor VIII observed. However, some individuals with undetectable levels of factor VIII antigen fail to generate factor VIII inhibitors, regardless of factor VIII exposure. Although these individuals would not be predicted to be tolerized to factor VIII, 70 to 80 percent of patients with baseline factor VIII levels of less than 1 percent do not develop an immune response to repeated dosing and are considered tolerized.13–16 For the 20 to 30 percent of patients who develop inhibitors, there are both genetic and nongenetic risk factors for inhibitor development. Patients with a positive family history of inhibitors, those who have large factor VIII gene deletions, and nonwhites have a higher risk of inhibitor development.17–20 The non–factor VIII genes—interleukin-10, tumor necrosis factor-α, and cytotoxic T-lymphocyte antigen 4–318 allele—are associated with inhibitor development.17–20 Nongenetic risk factors, such as infusing factor at the time of a “danger” signal (e.g., a surgical procedure), intense factor exposure, and prophylaxis versus no prophylaxis, also are associated with inhibitor development.16 Patients who receive factor at the time of a “danger” signal may experience sufficient tissue injury to provide the necessary immune activation through DAMPs. Furthermore, it remains possible that low-grade and potentially clinically undetectable infection may provide low levels of PAMPs that could likewise stimulate anti–factor VIII antibodies following factor VIII exposure. However, while PAMPs and/or DAMPs may provide the important immune activation signals,21,22 several studies using animal models suggest that significant factor VIII antibody development can occur in the absence of known tissue injury or DAMP exposure.23 Consistent with this, immune activation can occur in the apparent absence of DAMPs or PAMPs toward several model antigens.24 Unique B-cell populations, especially those in the spleen, can rapidly respond to bloodborne antigens in the absence of any identifiable PAMPs or tissue injury, suggesting that these cells may be uniquely poised to respond to factor VIII.25 Consistent with this, in experimental models, splenectomy can significantly inhibit factor VIII inhibitor development following factor VIII exposure,26,27 suggesting that several of these unique B-cell populations may be involved in the development of factor VIII antibodies irrespective of DAMP or PAMP exposure.25,26
Although examples of antigens inducing B-cell activation in the absence of known DAMPs or PAMPs exist, most of these antigens require crosslinking of cell-surface B-cell receptors for efficient activation and therefore reflect highly repetitive antigenic structures.28 In contrast, factor VIII represents a soluble antigen with little inherent predicted crosslinking ability. Most soluble antigen of this type actually induce tolerance following injection, likely because of the inability of soluble monovalent antigens to adequately crosslink and thereby stimulate B-cell receptors. Although factor VIII can exist in a soluble, monovalent form, it remains possible that factor VIII may form complexes with higher-molecular-weight species and thus form a network of factor VIII antigens that may serve as a suitable substrate for efficient B-cell receptor crosslinking and subsequent activation. Consistent with this, induction of tolerance to factor VIII by exposure to high levels of factor VIII may partially reflect a saturation of sites for factor VIII complex formation,29 which may, in turn, result in B-cell exposure to high levels of soluble, monovalent factor VIII. However, if this occurs, studies suggest that it likely takes place independent of interactions with von Willebrand factor, the primary binding partner of factor VIII, or its own coagulant activity.30 Clearly, there is much more to learn regarding the immunologic factors responsible for factor VIII inhibitor development.
In contrast to generating alloantibodies following factor VIII infusion, some patients generate autoantibodies against factor VIII, which can result in acquired factor VIII deficiency. As coagulation typically occurs at sites of inflammation and injury where DAMPs presumably are generated, tolerance to factor VIII may unfortunately be lost in these settings. Additionally, nonproteolytic and proteolytic degradation of coagulation proteins potentially could present neoepitopes. However, the fact that the development of acquired factor VIII deficiency is rare (1.4 per million population) provides a testimony to the ability of the immune system to discriminate efficiently between infectious nonself and noninfectious self.12 Essentially, nothing is known about the breakdown of tolerance in patients who develop autoantibodies to coagulation factors.
Factor VIII inhibitors in congenital and acquired hemophilia nearly always consist of a polyclonal immunoglobulin (Ig) G population. Although IgG4 accounts for only 5 percent of the total IgG in normal plasma, it usually is a major, but not the sole, component of the anti–factor VIII antibody population.31 IgG4 antibodies do not fix complement, which has been cited as a reason that immune complex disease is not observed in factor VIII inhibitor patients. However, it is more likely that factor VIII simply is not present in sufficient quantity to form enough immune complex deposition to mediate tissue damage.
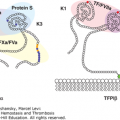
Factor VIII contains a sequence of domains designated A1-A2-B-ap-A3-C1-C2 (Chap. 13). During the activation of factor VIII by thrombin, the B and ap domains are released, producing an A1/A2/A3-C1-C2 activated factor VIII heterotrimer.32 Anti–factor VIII antibodies in both congenital and acquired hemophilia A inhibitor are primarily directed to the A2 and C2 domains, although antibodies to all domains have been described.33–35 The similarity in the properties of antibodies in congenital and acquired hemophilia, which represent very different immunologic settings, suggests that intrinsic structural features in the factor VIII molecule are an important determinant driving the immune response. Epitope spreading from a single “problem” epitope, which has been implicated in some autoantibody phenomena,36 does not appear to be a property of factor VIII inhibitors because anti–C2 antibodies can occur in the absence of anti–A2 antibodies and vice versa.
The only known biologic function of factor VIII is to become proteolytically activated and participate as a cofactor for factor IXa during intrinsic pathway factor X activation on phospholipid membranes. Theoretically, antibodies could inhibit factor VIII procoagulant function in several ways, including blocking the binding of factor VIIIa to factor IXa, factor X, or phospholipid, or by interfering with the proteolytic activation of factor VIII. Some anti–A2 antibodies map to a region bounded by Arg484-Ile50837 and inhibit activated factor VIII by blocking its ability to bind factor X.38 Anti–C2 antibodies bind to the NH2-terminal half of the C2 domain.39 Anti–C2 antibodies have been identified that inhibit the binding of activated factor VIII to phospholipid membranes,40 which is critical for its interaction with the platelet surfaces. However, the C2 domain also apparently contributes to the binding of factor VIII to its activators, thrombin and factor Xa.41–43 Consistent with this, anti–C2 inhibitors have been identified that block factor VIII activation.41,44
Factor VIII inhibitors also have been identified in approximately 20 percent of normal healthy donors.45 These inhibitors inhibit factor VIII activity in pooled normal plasma, but not autologous plasma, indicating that they are not autoantibodies, but rather alloantibodies directed against an unidentified polymorphism. Anti–factor VIII IgG also has been identified in all normal plasmas tested by affinity chromatography on immobilized factor VIII.46 The increased sensitivity of the method is a consequence of its ability to resolve anti–factor VIII antibodies from anti–anti–factor VIII idiotypic antibodies that also are present. Idiotypic regulation has been proposed as a mechanism for controlling autoantibody activity in vivo.47
Acquired hemophilia A patients usually present with spontaneous bleeding, which often is severe and life- or limb-threatening, although large cohort studies have shown that approximately 30 percent of patients do not require hemostatic management.2,48 Patients with acquired hemophilia are more likely to have a severe bleeding diathesis than congenital hemophilia A inhibitor patients.49 Additionally, in contrast to patients with congenital hemophilia A, hemarthrosis in these patients is rare. The reasons for these differences is puzzling, especially in light of the fact that the properties of factor VIII inhibitors in the two patient populations is similar. As noted above, inhibitors can block factor VIII function in several ways. Conceivably, unidentified mechanistic differences in inhibitor action account for the difference in clinical severity. Factor VIII inhibitors sometimes resolve spontaneously. However, it is not possible to predict in which subset of patients this will occur.
The new onset of an acquired bleeding disorder should immediately lead to screening tests that include an activated partial thromboplastin time (aPTT), a prothrombin time, and a platelet count. Patients with acquired hemophilia A have a prolonged aPTT resulting from decreased or absent factor VIII activity in the intrinsic pathway of blood coagulation. The autoantibody inhibits the factor VIII in the plasma from normal individuals, which forms the basis of the mixing study that is used to screen for inhibitors. The presence of a prolonged aPTT in a mixing study establishes the diagnosis of a circulating anticoagulant. Specific factor assays then are performed to determine whether a specific coagulation factor inhibitor or a lupus anticoagulant is present. The activity of other intrinsic pathway coagulation factors may be decreased in the presence of high titer factor VIII inhibitors. However, the levels of these factors normalize at increasing dilutions of patient plasma, whereas factor VIII activity remains decreased.
Once the identity of an inhibitor has been established, its titer is determined using the Bethesda assay.50 Inhibitors frequently take minutes to hours to maximally inhibit factor VIII. Therefore, dilutions of patient plasma are preincubated with normal plasma for 2 hours at 37°C. The inhibitor titer is defined as the dilution of patient plasma that produces 50 percent inhibition of the factor VIII activity and is expressed in Bethesda units per milliliter (BU/mL). Inhibitors are classified informally as low titer or high titer when the titers are less than 5 BU/mL or greater than 5 to 10 BU/mL, respectively. The Bethesda assay has been modified by the addition of 0.1 M imidazole, pH 7.4, and by diluting test plasma into factor VIII–deficient plasma during the preincubation phase to prevent assay variation resulting from pH changes and adsorptive losses of factor VIII.51 This “Nijmegen” modification of the Bethesda assay decreases false-positive low-titer inhibitors.52 Patients with acquired hemophilia often have measurable residual factor VIII activity. This activity may cause an underestimate of the inhibitory titer. Preanalytical heat treatment has been proposed as a simple way to denature factor VIII to allow for more accurate determination of titer in both patients with acquired hemophilia A and patients with congenital hemophilia A who may have infused factor VIII.53,54
Factor VIII inhibitors are classified based on the kinetics and extent of inactivation of factor VIII in plasma.55 Type I inhibitors follow second-order kinetics and inactivate factor VIII completely, which would be expected for a simple bimolecular antigen-antibody reaction. Type II inhibitors inactivate factor VIII incompletely and display more complex kinetics of inhibition. Hemophilia A inhibitor patients and acquired hemophilia A patients tend to have type I and type II inhibitors, respectively.56 However, the borderline between type I and type II inhibitors is not always clear and the distinction is not useful clinically. Additionally, in a recent observational study of patients with acquired hemophilia in the United Kingdom, factor VIII levels and inhibitor titers at presentation were not predictive of the severity of bleeding events. The median factor VIII level and inhibitory titers were nearly identical for patients with fatal bleeding events compared to those who did not require treatment for their bleeding symptoms.2
The severe bleeding that often is the presenting feature of this disorder requires urgent action to establish a diagnosis and initiate therapeutic measures. Ideally, this is carried out in a setting where factor VIII inhibitors can be identified and quantitated and where there is subspecialty expertise in the management of bleeding disorders. Invasive procedures should be performed only if absolutely necessary, and venipuncture should be kept to a minimum given the risk of significant bleeding.57
Treatment of patients with acquired hemophilia A depends on the inhibitor titer. Although no prospective trials are available, clinical experience indicates that patients with a factor VIII inhibitor titer of less than 5 BU/mL often are treated successfully with sufficient doses of recombinant or plasma-derived factor VIII to neutralize the inhibitor. Patients with titers between 5 and 10 BU/mL also may respond to factor VIII, whereas those with titers greater than 10 BU/mL generally do not respond. Formulas exist to calculate the amount of factor VIII needed to treat a patient, but these are rough estimates at best. The efficacy of factor VIII concentrates was lower than that of bypassing agents in a large registry study, which was likely secondary to challenges in appropriately dosing the factor VIII concentrate.48
Desmopressin can be administered by intravenous, subcutaneous, or intranasal routes and results in an increase in plasma von Willebrand factor levels and factor VIII activity.58 Its potential use is in patients with baseline factor VIII levels greater than 5 IU/dL and minor bleeding. However, like factor VIII concentrates, response is not predictable and close monitoring of hemostatic efficacy and factor VIII levels is needed.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


