Dosing regimen
Duration of infusion (h)
MIC (μg/mL)
Probability of patients achieving a target T > MIC of
30 %
35 %
40 %
45 %
500 mg every 8 h
1/2/3
1
100/100/100
100/100/100
100/100/100
99/100/100
500 mg every 8 h
1/2/3
2
100/100/100
99/100/100
77/100/100
25/90/100
1,000 mg every 12 h
4/5/6
4
100/100/100
100/100/100
92/100/100
23/96/100
1,000 mg every 8 h
1/2/3
4
100/100/100
99/100/100
77/100/100
25/90/100
1 g/2 g/3 g every 24 h
24
4
0/98/100
0/98/100
0/98/100
0/98/100
1,000 mg every 8 h
1/2/3
8
100/100/100
100/100/99
84/99/99
26/90/95
1 g/2 g/3 g every 24 h
24
8
0/0/46
0/0/46
0/0/46
0/0/46
Table 14.2
Commonly recommended standard and high doses (in adult patients with normal renal function) per day for selected betalactams and their plasma half-life and chemical stability at room temperature which are relevant for extended infusion regimens
Standard dose per day | High dose per day | Half-life | Chemical stability (at room temperature) | Commonly used extended infusion times | |
|---|---|---|---|---|---|
Flucloxacillin | 2 g every 8 h | 3 g every 6 h | 1 h | 24 h | 4–6 h |
Aztreonam | 1 g every 12 h | 2 g every 6 h | 1.5 h | >24 h | 4–8 h |
Piperacillin-tazobactam | 3/0.375 g every 6 h | 4/0.5 g every 6 h | 1 h | 24 h | 4–6 h |
Ceftazidime | 1 g every 12 h | 2 g every 8 h | 2 h | 24 h | 4–8 h |
Cefepime | 1 g every 12 h | 2 g every 8 h | 2 h | 24 h | 4–8 h |
Imipenem | 0.5 g every 8 h | 1 g every 6 h | 1 h | 3–4 h | 3–4 h |
Meropenem | 0.5 g every 8 h | 2 g every 8 h | 1 h | 4–8 h | 3–4 h |
Doripenem | 0.5 g every 8 h | 0.5 g every 6 h | 1 h | 4 h | 3–4 h |
Most betalactams are hydrophilic antibiotics with small distribution volumes similar to extracellular water. Tissue concentrations are usually not higher than plasma concentrations and largely depend on protein binding. Most studies in the field indicate that the higher the protein binding is, the lower is the tissue concentration. The ability to penetrate into various deep compartments differs for specific betalactams, depending on additional characteristics, notably physicochemical properties in relationship to tissue/cell binding and to drug carriers/active transporters across anatomical barriers.
14.1.1 Piperacillin-Tazobactam
Piperacillin is an ureidopenicillin which, in the absence of a betalactamase inhibitor, is inactivated by most betalactamases which are now very common among various Enterobacteriaceae. The distribution of MICs of the drug alone and in combination with tazobactam for E. coli, K. pneumoniae, and P. aeruginosa is shown in Fig. 14.1. As can be seen, piperacillin alone has limited activity against both E. coli and K. pneumoniae. In the presence of tazobactam, the MICs of both E. coli and Klebsiella are shifted to the “susceptible” range, while the addition of tazobactam does less alter the distribution of piperacillin MICs against P. aeruginosa [35]. The activity of the drug combination is better than that of ticarcillin-clavulanic acid which is still marketed in many countries (Table 14.3). Most A. baumannii and Stenotrophomonas maltophilia, however, are resistant to piperacillin-tazobactam [35].
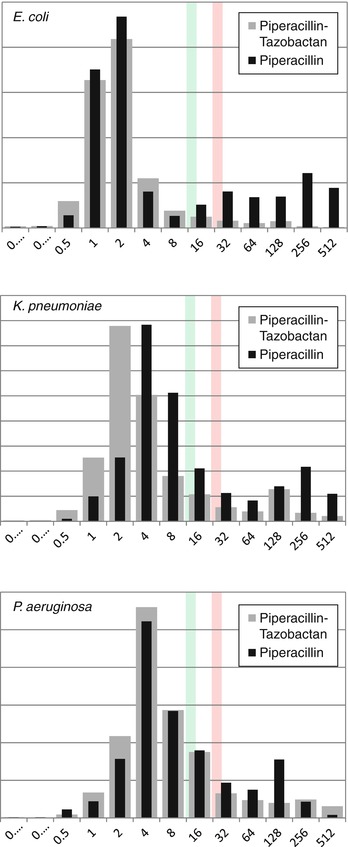
Fig. 14.1
The distribution of MICs of the piperacillin with and without tazobactam for E. coli, K. pneumoniae, and P. aeruginosa (Data are from EUCAST [35]). The green line indicates the breakpoint for susceptibility. The red line indicates the breakpoint for resistance
Table 14.3
Comparison of rates of resistance (in %) to betalactam versus betalactam/betalactamase inhibitor combinations according to EUCAST [35]
E. coli | K. pneumoniae | P. aeruginosa | |
|---|---|---|---|
Ampicillin | 43 % | 89 % | – |
Ampicillin-sulbactam | →34 % | →28 % | – |
Ticarcillin | 50 % | 97 % | 73 % |
Ticarcillin-clavulanic acid | →23 % | →22 % | →78 % |
Piperacillin | 30 % | 26 % | 24 % |
Piperacillin-tazobactam | →3 % | →10 % | →14 % |
Depending on the particular type(s) of extended-spectrum betalactamase (ESBL) enzyme(s), the drug is also quite active in vitro against ESBL-positive Enterobacteriaceae, and good clinical responses with the drug have been observed in bacteremic infection due to ESBL-positive E. coli if the MIC was <8 μg/mL [96]. Interestingly, in such cases amoxicillin-clavulanic acid (if tested susceptible) may be even more active due to the fact that clavulanic acid binds much more tightly to the ESBL enzyme than tazobactam (and sulbactam) – hence the designation “suicide” betalactamase inhibitor for clavulanic acid [31].
The antibacterial activity of piperacillin includes ampicillin-susceptible Enterococcus species, streptococci (including Streptococcus pneumoniae), and – if combined with tazobactam or other betalactamase inhibitors – many anaerobes and methicillin-susceptible staphylococci. For targeted therapy of proven staphylococcal infections, however, oxacillin and derivatives (in the case of methicillin-susceptible S. aureus) are superior.
Piperacillin-tazobactam is marketed as fixed combination formulation with 2/0.25 g, 3/0.375 g, and 4/0.5 g. In many countries a dose of 4/0.5 g every 6–8 h is the recommended daily dosage in adults with normal renal function. After a single dose of 4 g of piperacillin, peak plasma concentrations are in the range of 300–400 μg/mL. The half-life of piperacillin is approximately 1 h. The plasma concentration 6 h after short (~30 min) infusion is usually <2 μg/mL. The breakpoint for resistance according to EUCAST (European Committee on Antimicrobial Susceptibility Testing, www.eucast.org) is >16 μg/mL. The drug is stable for 24 h at room temperature [95, 112]. Extended infusion times including 24 h of continuous infusion are possible and safe. We recommend intermittent administration with extended infusion for 4–6 h.
The efficacy of initial monotherapy with piperacillin-tazobactam in patients with fever and neutropenia has been demonstrated in several clinical trials including a large double-blind trial from Italy in which the drug was compared with the initial combination of piperacillin-tazobactam plus amikacin [30].
14.1.2 Ceftazidime
Ceftazidime was the first antipseudomonal cephalosporin with excellent activity also against most Enterobacteriaceae. The drug was extensively evaluated in clinical trials in patients with fever and neutropenia [29, 36, 43, 49, 88, 89]. Typically, the MIC for susceptible P. aeruginosa is 1–4 μg/mL while the MIC for non-ESBL-positive E. coli and Klebsiella is in the order of 0.1–0.2 μg/mL [35]. The activity of ceftazidime against streptococci (including pneumococci) is somewhat lower than that of cefotaxime/ceftriaxone and of cefepime (MICs, ~0.25 versus <0.1 μg/mL). Similarly, the drug has more limited activity against methicillin-susceptible staphylococci compared with cefotaxime/ceftriaxone and cefepime (MICs, ~4 versus ~2 μg/mL) and is not recommended as targeted therapy in proven infections due to staphylococci or streptococci.
After intravenous infusion of 1 and 2 g doses of ceftazidime over 20–30 min in volunteers, peak plasma concentrations of ~70–100 and 120–180 μg/mL, respectively, are achieved. The half-life is approximately 2 h, and less than 10 % of the drug is protein bound. Elimination is almost completely by the renal route. Continuous infusion (6 g per day in patients with normal renal function) yields steady-state plasma concentrations of 30–50 μg/mL. Some centers use continuous infusion of 4 g per day (after an initial 2 g loading dose) [34, 88].
Pivotal trials of ceftazidime monotherapy in patients with fever and neutropenia have been published in the early to mid 1980s and established the efficacy and non-inferiority of ceftazidime versus cephalothin plus gentamicin plus carbenicillin, ceftazidime plus flucloxacillin, or piperacillin or azlocillin each plus an aminoglycoside [29, 36, 49, 89], and a large clinical trial reported equal efficacy of ceftazidime monotherapy compared with imipenem [43].
Today, the usefulness of the drug in particular for initial therapy has become limited primarily due to the emergence of ESBL-positive Enterobacteriacaeae [4, 74]. Using a breakpoint for resistance in Enterobacteriaceae of >4 μg/mL (EUCAST), approximately 60–70 % of ESBL-positive isolates will be resistant to ceftazidime (compared with 20–30 % resistance to piperacillin-tazobactam in such strains). Of note is that the breakpoint for resistance in P. aeruginosa was set at >8 μg/mL (dosage, 6 g per day) so that it is quite possible that ceftazidime at high dose and given by extended or continuous infusion will cover some more Enterobacteriaceae than EUCAST-defined in vitro resistance rates do suggest. Clinical experience with the drug in infection due to ESBL-positive Gram-negative bacteria is, however, limited.
Ceftazidime is currently developed in combination with a new betalactamase inhibitor, avibactam [116]. The potent in vitro activity of this new ceftazidime plus avibactam combination against Enterobacteriaceae producing betalactamases of Ambler class A, and more importantly class C and some class D, has been confirmed in vivo in murine pneumonia, bacteremia, and pyelonephritis models, and it is likely that clinical trials will show the usefulness of this new drug in patients with fever and neutropenia. Ceftazidime plus avibactam is a promising regimen for infections due to Klebsiella with carbapenem resistance due to KPC enzymes.
14.1.3 Cefepime
Cefepime was brought to market in the mid 1990s. The antimicrobial activity is similar to that of ceftazidime with somewhat lower MICs of cefepime for staphylococci and streptococci. As with ceftazidime, the MIC for susceptible P. aeruginosa is typically 1–4 μg/mL, while the MIC for non-ESBL-positive E. coli and Klebsiella is in the order of <0.1–0.2 μg/mL [35]. The drug is more stable than ceftazidime to AmpC-type betalactamases which are typically found in Enterobacter species.
There still are major discrepancies between the USA and Europe in setting clinical breakpoints for the drug. According to the so-called CLSI (Clinical and Laboratory Standards Institute, proposing breakpoints in the USA, www.clsi.org) standards, the breakpoint of cefepime for resistance in Enterobacteriaceae had earlier been set at >32 μg/mL, while according to EUCAST criteria, it is now >4 μg/mL. This has implications for estimating and interpreting susceptibility rates, particularly for ESBL-producing K. pneumoniae and E. cloacae. The rates of cefepime susceptibility for such strains may differ substantially, for example, 61 % (CLSI) versus 12 % (EUCAST) for K. pneumoniae or 58 % versus 32 % for E. cloacae, respectively [114]. Since the pharmacokinetic characteristics of cefepime are similar to those of ceftazidime and dosing is also similar for the two drugs, it may be more logical to apply breakpoints which are also similar. The most recent CLSI criteria revision (in 2014) consequently changed the breakpoint of cefepime for resistance in Enterobacteriaceae to >8 μg/mL (breakpoint for susceptibility, ≤2 μg/mL). In India, cefepime is also available as a fixed combination with sulbactam and with tazobactam – no clinical trials with those combinations have been reported as yet [109].
Cefepime was approved in 1997 for use as empiric monotherapy in febrile neutropenic patients in the USA. In 2007, a meta-analysis noted increased 30-day mortality associated with the use of cefepime versus other betalactams (risk ratio, 1.26) [115]. Among subcategories of patients, the increased mortality with cefepime was statistically significant only among neutropenic patients [87, 115]. The USA Food and Drug Administration reevaluated the data in collaboration with the drug sponsor that provided additional unpublished material and, 2 years later, concluded that the data would not confirm a higher rate of death in cefepime-treated patients [65]. There has still been some concern over cefepime’s safety, and there continue to appear reports about possibly dose-related neurotoxicity including nonconvulsive status epilepticus that can be severe and life-threatening [44, 62, 107].
14.1.4 Carbapenems
Imipenem was the first carbapenem introduced into the market [61]. It can be hydrolyzed in the mammalian kidney by an enzyme dubbed dihydropeptidase-I. This brush-border enzyme is inhibited by the co-drug cilastatin. With this imipenem/cilastatin combination, high urinary concentrations and recovery are obtained, and imipenem does not enter into the proximal tubular epithelium of the kidney and cannot be metabolized to a metabolite causing tubular necrosis.
Meropenem was the second carbapenem approved in both the USA and Europe and has been extensively studied in patients with fever and neutropenia. Meropenem is – besides piperacillin-tazobactam – now commonly used for initial empirical monotherapy in cancer patients [23, 39, 40, 43, 45, 77]. The advantage of meropenem is its broad-spectrum activity and excellent tolerability. Other carbapenems in the market include ertapenem, a carbapenem for single daily dosing that has no activity against P. aeruginosa; doripenem which is similar to meropenem [45]; and two other compounds only approved in Japan (panipenem and biapenem) [69].
The carbapenems are stable to most betalactamases including AmpC and ESBL enzymes. Resistance develops when bacteria acquire so-called metallobetalactamases or Ambler class A-type KPC enzymes that are capable of degrading carbapenems or when changes in membrane permeability arise usually as a result of loss of specific outer membrane porins. The activity of carbapenems so far remains excellent against wild-type Enterobacteriaceae (MIC in E. coli and K. pneumoniae, 0.01–0.06 μg/mL) with few exceptions. Species of Proteus, Morganella, and Providencia often are marginally susceptible or (low-level) resistant to imipenem and ertapenem [35].
Most isolates of P. aeruginosa and of A. baumannii show an MIC of meropenem in the range of 0.2–2 μg/mL [35]. Carbapenems are active against most clinically relevant anaerobes. They are poorly active against enterococci (typical MIC of meropenem, 2–16 μg/mL) and inactive at clinically achievable concentrations against S. maltophilia (typical MIC of meropenem, >32 μg/mL). Listeria are surprisingly susceptible to meropenem and imipenem which also have activity against Nocardia species and (extracellular) mycobacteria including multidrug-resistant M. tuberculosis.
Imipenem, meropenem, and doripenem have in vivo half-lives of approximately 1 h, while ertapenem has a half-life of approximately 4 h. Imipenem reaches higher CSF concentrations than meropenem, but meropenem is usually preferred for the treatment of CNS infections due to its lower incidence of seizures. The carbapenems are chemically less stable at room temperature than ceftazidime and piperacillin-tazobactam, in particular in concentrated (>4 g/100 mL) aqueous solution, and the rate of inactivation within 24 h can exceed 50 % [7, 112]. Although there is some experience with longer infusion times [68], we believe extended infusion regimens >4 h cannot currently be recommended.
Meropenem has extensively been evaluated in patients with fever and neutropenia. In the largest study (already published in 1996 and) involving more than 1,000 subjects [23], meropenem was associated with a slightly higher clinical response than ceftazidime plus amikacin (56 % versus 52 %, respectively). In a second large trial with 411 adult cancer patients, a similar response rate was observed with meropenem (54 % versus 44 % in patients receiving ceftazidime monotherapy, respectively) [39]. In children, a response rate of 56 % was reported (versus 40 % in pediatric patients receiving ceftazidime monotherapy, respectively) [40]. Some trials noted diarrhea with or without Clostridium difficile infection as a frequent significant adverse event, but carbapenems were otherwise usually well tolerated. There are very limited data on head-to-head comparisons of carbapenems for empirical therapy in febrile neutropenia.
The breakpoints of meropenem for resistance in Gram-negative bacteria are >8 μg/mL (EUCAST) and between >2 and >8 μg/mL (last CLSI revision, depending on whether Enterobacteriaceae, P. aeruginosa, or A. baumannii is tested) (Table 14.4). Such “more or less” carbapenem-resistant Gram-negative microorganisms show variable MICs ranging from 4 to >64 μg/mL. Importantly, meropenem used at high dosage (3 × 2 g by extended infusion instead of 3 × 1 g) is likely to cover microorganisms with an MIC of 16 and 32 μg/mL with a T>MIC of 50–60 % and 20–30 %, respectively [58]. The safety of such a dosage regimen has recently been shown in a comparative clinical trial [22]. Such a regimen can be considered as important adjunct and combination therapy partner to effectively treat infections due to carbapenemase-producing organisms [4, 25, 74].
Table 14.4
The breakpoints of meropenem for susceptibility and resistance (μg/mL) for Gram-negative bacteria according to EUCAST and CLSI
CLSI | EUCAST | ||||
|---|---|---|---|---|---|
S | R | S | R | ||
Meropenem | Enterobacteriaceae | ≤1 | >2 | ≤2 | >8 |
Acinetobacter | ≤4 | >8 | ≤2 | >8 | |
P. aeruginosa | ≤2 | >16 | ≤2 | >8 | |
Imipenem | Enterobacteriaceae | ≤1 | >2 | ≤2 | >8 |
Acinetobacter | ≤4 | >8 | ≤2 | >8 | |
P. aeruginosa | ≤2 | >16 | ≤4 | >8 | |
14.1.5 Other Betalactams
Among the various other betalactams commonly used and potentially useful in patients with fever and neutropenia are oxacillin and its derivatives, amoxicillin-clavulanic acid, ceftriaxone, and aztreonam. Cefixime has also been evaluated in step-down therapy in a very limited number of children with low-risk febrile neutropenia without documented infection. There is insufficient information to consider this drug as a suitable alternative to amoxicillin-clavulanic acid in patients with fever and neutropenia.
14.1.5.1 Oxacillin
The oxacillin derivatives cloxacillin, dicloxacillin, and flucloxacillin (like nafcillin) are narrow-spectrum penicillins with activity against methicillin-susceptible staphylococci and many streptococci, but no relevant antimicrobial activity against enterococci and Listeria. Oxacillin and its derivatives are important drugs in severe staphylococcal infection and are considered to be superior to vancomycin and also to cefazolin and to cefuroxime in patients with bacteremic S. aureus infection. The MIC of oxacillin in staphylococci is typically ~0.25–0.5 μg/mL [35]. Based on population pharmacokinetic data, a daily dose of 6 g given as 2 g by 4 h extended infusion every 8 h is likely to provide an fT > MIC of 50 % at an MIC of 0.75–1 μg/mL [71]. Short infusion regimes will require higher daily dosages to maintain high probabilities of response. These data have been the basis for recommendations of 2 g given every 6–8 h daily for serious staphylococcal infection. Higher dosages (Table 14.2) may be required in staphylococcal endocarditis and central nervous system infection.
The most critical adverse event apart from rash associated with oxacillin and its derivatives is cholestatic jaundice and hepatitis. Risk factors appear to be older age, preexisting hepatic impairment, long-term use (>14 days), and probably the daily dose. It is unknown whether continuous or extended infusion regimens can lower the risk of hepatotoxicity. Flucloxacillin is stable at room temperature for at least 24 h.
14.1.5.2 Amoxicillin-Clavulanic Acid
As an oral drug, amoxicillin-clavulanic acid is commonly used together with ciprofloxacin for therapy in low-risk febrile neutropenia patients [42]. Amoxicillin is well absorbed. Its oral bioavailability is approximately 70 %. A dose of 500 mg yields a mean peak plasma concentration of ~7–10 μg/mL [41, 94]. The usual oral dose of 500 mg (together with 125 mg of clavulanic acid) every 8 h provides a ~55 % or greater T > MIC for Haemophilus and streptococci incl. pneumococci [6]. Almost similar results (T > MIC [1 μg/mL], 44 %) are obtained with the 875/125 mg tablets given every 12 h [5]. Elimination of amoxicillin is predominantly by the renal route (52 %), and high concentrations are achieved in the urine sufficient to treat urinary tract infection (including those due to Gram-negative bacteria susceptible at the breakpoint of 8 μg/mL) [41, 94]. Parenteral therapy with higher doses is needed to treat systemic infections due to susceptible Gram-negative bacteria. In some countries, organisms with an MIC of 1–8 μg/mL are therefore categorized as intermediate susceptible. Resistance in E. coli is not uncommon and is primarily due to enzymatic inactivation by OXA-1- or inhibitor-resistant TEM (IRT) enzymes and chromosomal AmpC hyperproduction [97].
Clavulanic acid is extensively metabolized and eliminated in urine and feces. Adverse effects include rash, gastrointestinal disturbances, and – rarely – cholestatic hepatitis usually associated with clavulanic acid rather than with amoxicillin. Age, preexisting liver disease, therapy duration, and probably genetic factors play a role in the risk for drug-induced hepatitis [106].
14.1.5.3 Ceftriaxone
Ceftriaxone has become a most popular parenteral broad-spectrum cephalosporin because of its long half-life and the ease of the corresponding once daily dosing. It is a third-generation cephalosporin with antimicrobial activity very similar to that of cefotaxime. The usual dose in adults for infections outside the brain is 2 g every 24 h. This dosing regimen yields ~100 % fT > MIC (≤1 μg/mL), thus providing excellent in vivo activity against streptococci, Haemophilus, and wild-type Enterobacteriaceae. It has no relevant activity against P. aeruginosa (MIC typically >8 μg/mL). The breakpoint for resistance has been set at >2 μg/mL (EUCAST). The drug is very well tolerated. Gastrointestinal disturbance may occur, and Clostridium difficile infections are more common than with most other broad-spectrum cephalosporins due to its substantial effects on the intestinal microflora.
14.1.5.4 Ceftaroline and Ceftobiprole
Both compounds are broad-spectrum cephalosporins approved for the treatment of skin and soft tissue infection and pneumonia. The two drugs have enhanced activity against MRSA and in the future might become interesting treatment options in febrile neutropenia. There is increasing experience with them in salvage therapy for MRSA infection [18].
14.1.5.5 Aztreonam
Aztreonam is a monobactam available for clinical use since the 1980s. The antimicrobial activity comprises exclusively Gram-negative bacteria. Wild-type E. coli and Klebsiella species are highly susceptible with MICs typically <0.1 μ/mL [35]. Aztreonam is moderately active against P. aeruginosa (MIC, ~4 μg/mL) and has no clinically relevant activity against Acinetobacter and Stenotrophomonas [35]. The MIC distribution for E. coli, P. aeruginosa, and A. baumannii is shown in Fig. 14.2. Aztreonam is not inactivated by Ambler class B betalactamases (metalloenzymes) and represents an important therapeutic option in infections due to P. aeruginosa with metallobetalactamase-associated carbapenem resistance. In some countries, the drug (as aztreonam-lysine with a dose of 75 mg administered every 8 h using an ultrasonic nebulizer) has been approved for inhalation [92].
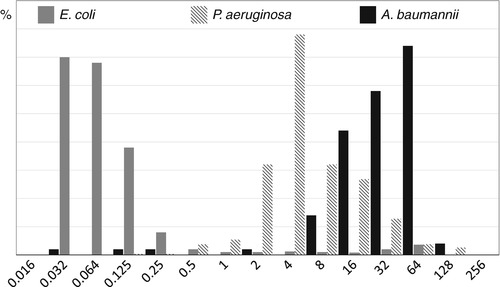
Fig. 14.2
The in vitro activity of aztreonam against E. coli, K. pneumoniae, and P. aeruginosa (Data are from EUCAST [35]). Aztreonam is moderately active against P. aeruginosa (MIC, ~4 μg/mL) and has no clinically relevant activity against Acinetobacter
Recommended doses for an adult patient with normal renal function are 1–2 g every 6–8 h. Monte Carlo simulations with a target of an fT > MIC of 50–60 % at a dose of 1 g (infused over 1 h) every 8 h indicate a clinical breakpoint of 1–4 μg/mL. Thus, infections due to susceptible E. coli do not require high-dose therapy. The drug can be given as continuous infusion (24 h). Continuous infusion of 6–8 g per day produces plasma concentrations of ~40–50 μg/mL [16]. Such a regimen should be sufficient to treat infection due to low-level resistant P. aeruginosa.
The addition of avibactam to aztreonam is currently being evaluated in clinical trials [24]. The advantage of such a drug combination would be the restored activity of aztreonam against ESBL-positive organisms and its intrinsic activity in organisms with metalloenzyme-associated carbapenem resistance.
14.2 Aminoglycosides
In view of many clinical trials establishing the noninferiority of betalactam monotherapy for fever and neutropenia [86], many clinicians now limit the use of aminoglycosides in these and other critically ill patients to an initial, often single-shot therapy in patients with severe sepsis/septic shock or recommend their use only for targeted combination therapy for infection due to otherwise difficult-to-treat or resistant organisms [13, 79, 82]. This strategy may change in the future if betalactam monotherapy becomes unreliable due to emerging resistance, and it is conceivable that there will be an increasing need to reinitiate initial empirical combination therapies with short courses of aminoglycosides.
The most commonly used aminoglycosides for systemic use are gentamicin, tobramycin, and amikacin. Other aminoglycosides such as netilmicin, isepamicin, and arbekacin are uncommonly used and/or not available in many countries [91]. New developments include plazomicin (a “neoglycoside”) which is stable against common aminoglycoside-inactivating enzymes [117].
The older aminoglycosides are most active in many Gram-negative bacteria [35]. Wild-type E. coli and Klebsiella exhibit gentamicin and tobramycin MICs in the range of 0.5–1 μg/mL and amikacin MICs in the range of 2–4 μg/mL. Aminoglycosides show a concentration-dependent bactericidal activity in Gram-negative organisms and an interesting so-called post-antibiotic effect that is also dependent on the ratio of drug concentration versus MIC and can last several hours [9]. Additional doses during this phase do not produce additional bacterial killing and the killing efficacy is reduced for subsequent repeated exposures (so-called adaptive resistance) compared to the initial exposure. Once daily dosing is therefore advantageous despite the relatively short half-life of the aminoglycosides of 2–3 h [9, 54, 79, 80, 83].
Fortunately, extended-interval dosing is also advantageous regarding the typical adverse effects of aminoglycosides, nephrotoxicity, and ototoxicity [9]. The toxic effects require drug uptake and binding to the brush borders of renal cells and to the cochlea and vestibular membranes. Binding to these membranes, however, demonstrates saturable kinetics and is less efficient at high intermittent drug levels. The half-life of aminoglycosides in the renal cortex is approximately 100 h, leading to renal accumulation of the drug and toxicity. In clinical trials, the advantages of once-daily dosing compared to two- to three-times daily administration has not been consistently shown, in part because often very potent betalactam combination therapy partners were used and treatment durations often were too short to fully assess differences in toxicity. Nevertheless, once-daily dosing of aminoglycosides is now recommended for most indications including treatment of patients with fever and neutropenia if betalactam monotherapy is unreliable.
There has been controversy about the relative risks of nephrotoxicity for gentamicin versus tobramycin (and netilmicin), respectively. In various animal models, gentamicin was more nephrotoxic than tobramycin if given at identical doses, and netilmicin at the same dose, in turn, was less nephrotoxic than tobramycin. Clinical studies in humans also provided some evidence for the greater potential of gentamicin to cause nephrotoxicity [38, 102]. Accordingly, gentamicin often is limited to combination therapy of enterococcal or streptococcal endocarditis where only low doses are required for synergistic action with penicillin. In patients with Gram-negative infection or suspected Gram-negative infection such as patients with fever and neutropenia (or urinary tract and abdominal sepsis and nosocomial pneumonia), higher doses are useful that make tobramycin, netilmicin, or amikacin drugs of choice.
Table 14.5 shows salient features of aminoglycoside therapy today. It should be noted that resistance rates may vary substantially between hospitals and regions depending among other things on what is the proportion of cephalosporin-resistant Enterobacteriaceae and carbapenem-resistant P. aeruginosa (which are often coresistant in particular against gentamcin and tobramycin) and on which aminoglycoside is being used most commonly.
Table 14.5
Salient features of commonly used aminoglycosides
Drug | Typical MIC (μg/mL) in wild-type cells | Break-points for resistance (μg/mL) | Resistance rates | Daily dose (per body weight) in subjects with normal renal function | Post-infusion plasma drug levels (μg/mL) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
E. coli | K. pneumoniae | P. aeruginosa | E. coli | K. pneumoniae | P. aeruginosa | Mean peak levels | Target levels at 8–12 h | Target trough levels (24 h) | |||
Gentamicin | 0.25–1 | 0.25–1 | 1–4 | >4 | 5 % | 11 % | 15 % | 1 × 4 mg/kg | 10–20 | <5 | <1 |
Tobramycin | 0.5–2 | 0.5–1 | 0.5–1 | >4 | 4 %
Related posts: Consequences from Specific Treatment Modalities
Fever of Unknown Origin: Treatment According to Risk Assessment
Antifungal Agents
Gastrointestinal Complications Consequences from Specific Treatment Modalities
Fever of Unknown Origin: Treatment According to Risk Assessment
Antifungal Agents
Gastrointestinal Complications
 The Diagnostic Approach to the Febrile Neutropenic Patient: Clinical Considerations The Diagnostic Approach to the Febrile Neutropenic Patient: Clinical Considerations
 Diagnostic Radiology in Hematological Patients with Febrile Neutropenia Diagnostic Radiology in Hematological Patients with Febrile Neutropenia
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||||||