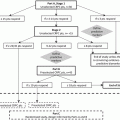
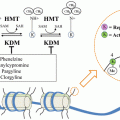
Fig. 15.1
TGF-β transforming growth factor-β; HGF hepatocyte growth factor; VEGF vascular endothelial growth factor; PLGF placental growth factor; Ang angiopoietin; PDGF platelet-derived growth factor; FGF fibroblast growth factor
Although there are many signaling pathways that direct angiogenic processes in both healthy and cancerous tissues (Fig. 15.1), the VEGF pathway has become the hallmark. Its downstream targets have been the best characterized, and it has received the most focus in the development of anti-angiogenic therapies.
VEGF is actually a family of seven related growth factors (VEGF-A through -E and placental growth factor [PLGF] 1 and 2), each with varying affinities for the several VEGF receptors. VEGF-A, originally known as vascular permeability factor (VPF), is a central mediator of angiogenesis, and has become a leading therapeutic target. Indeed, in the medical literature “VEGF” is used as synonymous with “VEGF-A.”
The secreted VEGF proteins bind to three known receptor tyrosine kinases: VEGFR-1 (Flt-1), VEGFR-2 (KDR/Flk-1), and VEGFR-3 (Flt-4). It is the action of VEGF-2, particularly upon binding the VEGF-A ligand, that has been the best studied in the context of anti-angiogenic therapy [39]. VEGFR-1 and VEGFR-2 expression is restricted largely to vascular endothelial cells, and their actions are relatively specific for this tissue. VEGFR-3, however, is restricted to lymphatic endothelial cells [40], and is felt to play a more important role in lymphangiogenesis than angiogenesis [41], though it has been shown to activate the Ras pathway similarly to VEGFR-2. Furthermore, VEGFR-3 does not bind VEGF-A, but rather VEGF-C and -D. The function of VEGFR-1 is not yet well understood, though it appears to play a role in monocyte migration, and a soluble version of the extracellular VEGFR-1 domain may be a negative regulator of angiogenesis during embryogenesis by acting as a “trap” for VEGF-A ligand [41].
Ligand binding results in homodimerization of VEGFR-2, leading to the activation of multiple intracellular pathways that upregulate expression of genes promoting the growth, survival, and migration of vascular endothelial cells. Activation of the protein kinase C (PKC)–Raf–MAP/ERK kinase (MEK)–MAPK/ERK pathway promotes endothelial cell growth. VEGFR-2 also activates the phosphatidylinositol 3-kinase (PI3K)–Akt/PKB pathway, which upregulates mTOR, a central driver of cellular proliferation and survival. Additionally, Akt/PKB also results in the phosphorylation of endothelial nitric oxide synthase (eNOS), increasing nitric oxide production and therefore increasing endothelial permeability [42]. Finally, by activating the focal adhesion kinase (FAK, also known as protein tyrosine kinase 2 or PTK2)–paxillin pathway, VEGFR-2 directs the migration and reorganization of endothelial cells to form new vessels [43].
Angiopoietin
Angiopoietin-mediated signaling via the TIE receptor tyrosine kinases is important for both embryonic angiogenesis and adult vascular homeostasis [44]. There is significant functional overlap between angiopoietin and VEGF, in terms of the downstream signals that each activates. Like VEGF-A signaling via VEGFR-2, angiopoietin signaling activates the Raf–MEK–MAPK pathway promoting endothelial cell growth [45]. It also activates the PI3K–Akt pathway, with similar upregulation of mTOR and eNOS, promoting vascular cell survival, proliferation, and permeability [45]. NF-kB is also activated, regulating inflammation [45].
There are two receptors for the angiopoietin family—TIE-1 and TIE-2. TIE-1 is poorly understood; it has no known ligand [46], but clearly plays some important role in embryogenesis, as knockout results in an embryonic lethal phenotype [47]. Additionally, the interaction between the angiopoietin family (Ang 1–4) and TIE-2 is complex. Ang-1 is a TIE-2 agonist. Ang-2, however, has a variable effect on TIE-2 that appears to be environment-dependent, serving as an antagonist in the absence of VEGF-A but as an agonist in the presence of VEGF-A [44]. Ang-3 and Ang-4 have limited homology to Ang-1 and Ang-2; their effects on TIE-2 and downstream angiogenesis pathways seem to be variable and dependent on the context [45].
Platelet-Derived Growth Factor
The platelet-derived growth factor (PDGF) family includes four peptides, A through D, which combine by homo- and hetero-dimerization to form a total of five functional, dimeric ligands: PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC, and PDGF-DD [48]. PDGF-mediated signaling has been shown to stimulate angiogenesis [49]. The transmembrane, tyrosine kinase receptors that bind the PDGF dimers are constituted from the PDGFR-α and PDGFR-β molecules, and include PDGFR-αα, PDGFR-αβ, and PDGFR-ββ. PDGF-BB is the only form of the protein that is capable of binding all three PDGFR dimers [48], a capability that may have functional significance as PDGF-BB appears to have greater angiogenic potency that the other PDGF dimers [50].
Like both VEGF and angiopoietin, PDGF signaling is also capable of activating both the Raf–MEK–MAPK and the PI3K–Akt pathways controlling cell proliferation and survival [51]. Additionally, PDGF also influences the NF-kB and Notch pathways, which, among other effects, increase cellular production of VEGF as well as matrix metalloproteinase 9 (MMP-9), promoting angiogenesis as well as invasion and metastasis [52]. Though there is a large degree of overlap in the downstream signaling cascades by which VEGF and PDGF act, PDGF-mediated signaling is capable of inducing angiogenesis independent of VEGF [53], highlighting its importance in this process and its potential as a therapeutic target.
Fibroblast Growth Factor
The FGF signaling system includes over 20 growth factors and four known FGF receptors (FGFR 1 through 4). Of this large family, FGF2 is the best-understood growth factor and has become the prototype for studying FGF signaling. FGFR1 is the primary receptor expressed on endothelial cells and mediates the growth, migration, and tubular morphogenesis of these cells, while FGFR2 primarily drives cell motility [54]. The FGF pathways are dependent on heparan sulfate proteoglycans, which act as coreceptors during FGF-FGFR binding [55].
FGF binding to FGFR activates the Ras–MEK–MAPK growth-regulating pathway. FGF signaling has also been observed to activate PI3K–Akt pathway, though this activity has been demonstrated only in non-vascular cells and its importance to FGF-mediated angiogenesis is unclear [54]. FGF also activates the p38 MAP kinase pathway, although this appears to down-regulate FGF-mediated angiogenesis, serving as a negative feedback mechanism [54]. As is the case for most of the angiogenesis pathways, FGF and FGFR are overexpressed in tumors [55]. There is thought to be a synergistic effect between FGF and VEGF [55] as well as FGF and PDGF [48] in the context of tumor neovascularization. FGF signaling may plan additional roles in prostate cancer specifically, as progressive expression of particular splice variants of FGFR2 has been associated with the development of androgen resistance in human prostate cancer tumor models [56].
Transforming Growth Factor-β
The transforming growth factor (TGF) ligand exists in three isoforms, TGF-β1 through 3. There are three classes of TGF-β receptors, types I–III, each of which has a distinct function in TGF-β signaling. Type III receptors do not directly engage downstream signaling cascades, but are thought to facilitate TGF-β binding to type II receptors; type II receptors then recruit and phosphorylate type I receptors, which in turn activate a group of transcription factors known as SMADs. Believed to be specific to TFG-β signaling, activated SMADs translocate to the nucleus and regulate transcription of genes involved in proliferation, differentiation, and angiogenesis [44].
The role of TGF-β in cancer is complex. SMAD-mediated signaling is generally recognized as apoptosis-inducing, which would have the expected effect of suppressing tumor growth. Indeed, there is some evidence to suggest that TGF-β functions as a tumor suppressor in early oncogenesis [57]. However, TGF-β has many other actions outside of the SMAD pathway, including activation of the Ras-Raf-MEK-MAPK and PI3K-Akt cascades that seem to serve as the “final common pathway” for many of the angiogenesis-inducing growth factors [58, 59]. Interestingly, tumorigenesis is often marked by both increased expression of TGF-β and loss-of-function mutations in the SMAD proteins [57], a sequence of events that may result in the preservation of the oncogenic effects of TGF-β within the tumor while avoiding its anti-proliferative functions.
Hepatocyte Growth Factor
Hepatocyte growth factor (HGF), also known as scatter factor (SF), controls a diversity of cellular processes. Its receptor is the transmembrane tyrosine kinase MET (c-MET, HGF receptor). Ligand binding induces MET homodimerization and autophosphorylation, activating the intracellular domain. HGF–MET signaled is primarily mediated by downstream activation of the Ras–Raf–MEK–MAPK and the PI3K–Akt pathways [60].
HGF has long been known to stimulate angiogenesis in vitro [61] and in vivo [62]. HGF–MET signaling may produce this effect via indirect action on other pathways. Specifically, HGF signaling leads to the inhibition of thrombospondin 1, a negative regulator of angiogenesis; HGF also induces VEGFA production, thereby upregulating one of the most potent angiogenic pathways [63].
Additionally, HGF–MET signaling is one of the pathways most clearly associated with the process of epithelial–mesenchymal transition (EMT). Normal EMT during embryogenesis allows for the migration of precursor cells, and is dependent on HGF–MET [60]. Similarly, EMT in the context of malignancy has been proposed as a central process in the development of metastatic capability [64]. The role of HGF–MET signaling has been well documented in many malignancies, including hepatocellular carcinoma [65], where the inhibition of HGF–MET–mediated EMT has been proposed as a possible mechanism of action of sorafenib in the treatment of this malignancy [66]. EMT in prostate cancer progression is understood to be induced by local factors in the tumor microenvironment, where it has been demonstrated to promote cell migration, invasion, and survival [64].
Specific Angiogenic Factors in Prostate Cancer
The biochemical mapping of angiogenesis pathways has also led to an increasing understanding of which signaling factors and pathways are involved in prostate cancer specifically. As in other cancers, VEGF appears to be one of the central players in prostate cancer angiogenesis. Beginning in the year 1997, immunohistochemical analysis of prostate tumors found high levels of VEGF expression in diseased tissue, compared with little or no expression in the normal prostate [67, 68]. Additional in vivo studies would confirm this result, and also demonstrate the absence of VEGF expression in benign prostatic hypertrophy [69]. Coexpression of the VEGF and VEGF receptor in prostate cancer microvasculature has also been reported [70].
VEGF expression in prostate cancer has since been correlated not only with angiogenesis, but also with outcomes. VEGF expression was found to correlate with risk of progression in one cohort [71]. Microvascular density in prostate cancer has also been associated with higher Gleason score and pathologic stage [72]. Findings such as these have led to the proposed use of angiogenesis as an additional prognostic variable in the examination of biopsy specimens. Furthermore, VEGF may have additional use as a circulating biomarker. High urine levels of VEGF may be an independent predictor of survival in the setting of castrate-resistant prostate cancer (CRPC); [73] though this correlation has not held up in other studies [74]. Elevated plasma VEGF levels have also correlated with poor outcomes [75]. These findings support a pathophysiologic role for VEGF in prostate cancer, and suggest that it may be a valid chemotherapeutic target.
Other angiogenic pathways are also important in prostate cancer. Introduction of neutralizing antibodies to the proteins secreted by cultured prostate cancer cells has found that FGF-2, in addition to VEGF, is partially responsible for the angiogenesis-inducing capability of these cells [76]. Immunohistochemical analysis of prostate tumors also demonstrates the activity of the FGF family, showing the coexpression of FGF-2 and the FGF receptor in diseased, but not normal, prostate tissue [77]. TGF-β overexpression has been linked not only with increased angiogenesis, but also with metastasis and poor patient outcomes [78]. Angiopoietin-2 also appears to play a role, as inhibition of its activity in a mouse prostate tumor model reduces microvascular density, cell proliferation, and serum PSA, with prolongation in survival [79]. Inhibition of angiopoietin-2 also increases expression of hypoxia-inducible factor-1α (HIF-1α) RNA, suggesting that the reduction in microvascular density results in ischemia within tumor tissue.
Intriguingly, the effects of androgen signaling, a central driver of prostate cancer growth, may be due in part to its stimulation of angiogenesis. In vitro assays have shown an increase in expression of both VEGF and the VEGF receptor in response to androgens [80]. This functional overlap suggests the potential for a synergistic effect from co-suppression of androgen and angiogenesis signaling pathways.
Potential Benefits of Anti-angiogenic Therapy in Combination Regimens
In prostate as well as other cancers, anti-angiogenic therapy has the potential to augment the effectiveness of other, traditional chemotherapeutics. While this effect is not completely understood, several potential mechanisms have been proposed. Tumor tissue has been described as having elevated interstitial fluid pressure, preventing adequate diffusion of pharmacologic agents; anti-angiogenic agents—specifically bevacizumab [81]—can act to reduce the intra-tumor hydrostatic pressure, thereby increasing drug delivery [82]. This effect may be mediated by promoting the normalization of the tumor microvasculature [83], again emphasizing the importance on the tumor microenvironment in determining its behavior. Normalization of the microvasculature may also serve to increase delivery of cytotoxic agents by decreasing vascular permeability and reducing the non-therapeutic extravasation of drug into the interstitial space [84]. Anti-angiogenic agents may also slow tumor growth by inhibiting autocrine and paracrine signaling [85]. Additionally, traditional chemotherapeutics and anti-angiogenic agents may produce combined effects simply by acting independently on different signaling pathways [85].
With regard to prostate cancer specifically, it is known that androgen signaling promotes tumor growth in part by stimulating angiogenesis. Introduction of androgens increases VEGF and VEGF receptor expression in prostate model cells [80]. Conversely, androgen blockade has been demonstrated to reduce VEGF expression in in vivo prostate cancer models, with concomitant reductions in microvascular density and tumor burden [86]. Androgen deprivation also prevents the hypoxia-induced neovascularization response within prostate tumors [87]. Findings such as these raise the possibility of a synergistic effect from combined blockade of androgen and angiogenic pathways.
Androgen signaling produces angiogenesis, at least in part, via the hypoxia-induced factor (HIF) pathway. Indeed, one of the processes leading to castrate resistance is the intra-tumoral upregulation of HIF-1, allowing the tumor to respond to reduced levels of androgen and thus survive in an androgen-deprived environment. As many anti-angiogenic agents inhibit the HIF-1 pathway, they may block this resistance mechanism and thus potentiate the effect of androgen blockade [88].
Anti-angiogenic Therapy in Prostate Cancer: Clinical Applications
Targeting the VEGF Pathway in Prostate Cancer
Bevacizumab
Preclinical data on bevacizumab for prostate cancer showed successful suppression of angiogenesis and proliferation of prostate cancer cell lines in vitro [89]. However, early clinical trial data failed to show significant activity. In the first phase II results for bevacizumab in prostate cancer, from an open-label trial of 15 patients with CRPC receiving bevacizumab monotherapy reported in 2001, no patient achieved a complete or partial response (Table 15.1). The best response was a “possible mixed response,” occurring in three patients; no PSA responses >50 % were observed [90].
Table 15.1
Phase II and III trials of bevacizumab for prostate cancer with reported results
Study title | Phase | N | Primary end point and results | Year | Reference |
|---|---|---|---|---|---|
A phase II trial of humanized anti-vascular endothelial growth factor antibody for the treatment of androgen-independent prostate cancer | II | 15 | Objective tumor response, PSA response 0 % objective tumor response, 27 % had PSA decline of >50 % | 2001 | Reese et al. The Prostate Journal, 3: 65–70 |
Combination of bevacizumab and docetaxel in docetaxel-pretreated hormone-refractory prostate cancer | II | 20 | Objective tumor response, PSA response 37.5 % objective partial response, 55 % had PSA decline of >50 % | 2008 | Di Lorenzo et al. [91] |
A phase II study of estramustine, docetaxel, and bevacizumab in men with castrate-resistant prostate cancer: results of cancer and leukemia group B (CALGB) 9006 | II | 79 | Progression-free survival Median progression-free survival was 8 months. Median overall survival was 24 months | 2011 | Picus et al. [92] NCT00016107 |
Phase 2 study of neoadjuvant docetaxel plus bevacizumab in patients with high-risk localized prostate cancer: a prostate cancer clinical trials consortium trial | II | 41 | Partial response by endorectal MRI 29 % experienced >50 % reduction in tumor volume | 2012 | Ross et al. [93] NCT00321646 |
Combination immunotherapy with prostatic acid phosphatase pulsed antigen-presenting cells (provenge) plus bevacizumab in patients with serologic progression of prostate cancer after definitive local therapy | II | 22 | PSA response 5 % had PSA decline of >50 % | 2006 | Rini et al. [94] |
Randomized, double-blind, placebo-controlled phase III trial comparing docetaxel and prednisone with or without bevacizumab in men with metastatic castration-resistant prostate cancer: CALGB 90401 | III | 1,050 | Overall survival Overall survival was 22.6 months in the docetaxel + prednisone + bevacizumab group, compared to 21.5 months for the docetaxel + prednisone group (p = 0.181) | 2012 | Kelly et al. [95] NCT00110214 |
Phase II trial of bevacizumab, thalidomide, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer | II | 60 | PSA response 90 % had PSA decline of >50 % | 2010 | Ning et al. [98] NCT00091364 |
It took several years for interest in bevacizumab for prostate cancer to return, but eventually the research focus shifted to combination chemotherapy, with more success. In 2008, a phase II trial of combination bevacizumab plus docetaxel in 20 patients previously treated with docetaxel reported a >50 % PSA decline in 55 % of patients [91]. Thereafter, a larger phase II trial of bevacizumab in combination with docetaxel and estramustine for CRPC found more encouraging results, with 75 % of its 79 patients achieving a >50 % PSA response, and 59 % of those with measurable disease having a partial response [92]. More recently, bevacizumab has been combined with docetaxel in the neoadjuvant setting before prostatectomy for high-risk localized disease, achieving pre-surgery reductions in tumor volume and PSA, but not yet with long-term follow-up on surgical cure rates [93]. Bevacizumab has also been tested in combination with sipuleucel-T, though more data are needed to test the hypothesis that bevacizumab may potentiate the response to this and other immunotherapies [94].
Based on these phase II results, a placebo-controlled phase III trial randomizing CRPC patients to docetaxel and prednisone with or without bevacizumab was undertaken (CALGB 90401). 1,050 patients were enrolled. There was no difference in the primary endpoint of overall survival (OS) (22.6 months in the bevacizumab arm vs. 21.6 months in the placebo arm, p = 0.181), although secondary end points of median progression-free survival (PFS) (9.9 vs. 7.5 months, p < 0.0001), PSA response, and objective response favored the bevacizumab group. Additionally, bevacizumab appeared to increase toxicity, with an increase in treatment-related deaths (3.8 vs. 1.1 %) [95].
There were several reasons why the CALGB 90401 trial may have produced divergent results for PFS and OS, including a greater burden of comorbidities in the bevacizumab arm [96], as well as not continuing bevacizumab treatment beyond the completion of docetaxel therapy, resulting in shorter treatment durations [97]. In subgroup analysis, those patients with poor prognostic factors (such as elevated LDH and alkaline phosphatase) derived greater benefit from bevacizumab treatment. Additionally, anti-VEGF therapies such as bevacizumab may provide a far greater benefit to those patients whose tumors, and trials that are not enriched for such patients may be underpowered to detect this benefit. Analysis of other trial cohorts (CALGB 9480) has identified elevated plasma VEGF levels as an independent poor prognostic factor [75], suggesting the possibility of selecting for patients with VEGF-dependent biology in future trials.
Since then, combination therapy with bevacizumab for CRPC has taken new directions. A phase II trial of bevacizumab in combination with thalidomide, prednisone, and docetaxel achieved a 90 % rate of >50 % PSA reduction, though this combination was limited by toxicities such as bone marrow suppression [98]. A similar regimen of bevacizumab, lenalidomide, prednisone, and docetaxel is currently under investigation [NCT00942578]. Other ongoing clinical trials are expected to report for the first time on bevacizumab in combination with the mTOR inhibitors everolimus [NCT00574769] and temsirolimus [NCT01083368]. Also of notable interest due to the theoretical synergistic effect [88], several trials are underway to test combined androgen and angiogenesis suppression. One phase II trial aims to test androgen blockade with bicalutamide with or without bevacizumab as first-line therapy after PSA recurrence following prostatectomy [NCT00776594]. Another ongoing phase II trial will evaluate bevacizumab and androgen deprivation with docetaxel in a similar first-recurrence setting [NCT00658697]. However, without study designs enriching for patients with VEGF-dependent tumor biology, these trials are less likely to achieve new results that will significantly advance the field.
Aflibercept
Aflibercept also blocks the VEGF pathway, but by a different mechanism than bevacizumab; containing regions of the VEGF receptors 1 and 2, this protein acts as a soluble VEGF “trap.” Phase I studies were promising, showing signals of activity when used in combination with docetaxel for a variety of cancer types, including prostate cancer [99]. Based on these data, aflibercept was taken directly to the phase III stage, being tested with docetaxel and prednisone in the >1,200 patient VENICE trial [NCT00519285] (Table 15.1). In the VENICE trial, aflibercept did not improve the primary outcome of overall survival compared to placebo, and was associated with an increased risk of multiple toxicities [100]. Similarly to CALGB 90401, the VENICE trial did not enrich its study cohort for poor prognostic factors or elevated VEGF levels, leaving open the possibility that a clinically significant benefit may be possible in a selected cohort with these features. While trials continue to search for new applications for bevacizumab in the treatment of CRPC, the role of aflibercept in the future treatment of this disease remains unclear.
Immunomodulatory Agents: The Thalidomide Family
Thalidomide
Although its potential harms as a devastating teratogen were already known, thalidomide was also demonstrated to be a potent anti-angiogenic factor in the mid-1990s. By use of a rabbit cornea micropocket assay, thalidomide was shown to inhibit FGF-mediated angiogenesis [101]; subsequent research would also show its ability to prevent tumor growth in a rabbit model [102]. The mechanism of the thalidomides’ anti-angiogenic activity is not fully understood. Reduction in angiogenic factors such as VEGF and FGF has been observed, along with a pro-apoptotic effect [103]. Additionally, thalidomide has a significant immunomodulatory effect via inhibition of TNF-α, which is also felt to contribute to its anti-angiogenic capability [44].
Since the discovery of its anti-angiogenic properties, thalidomide and its derivatives have become the foundation of treatment for multiple myeloma, significantly improving outcomes in that disease. Applications for prostate cancer have begun to be explored, as well.
Phase II data for prostate cancer began to be reported in 2001 (Table 15.2). A 63-patient trial of thalidomide in combination with docetaxel for CRPC observed a >50 % PSA reduction in 18 % of patients, but the response rate did not appear to increase with higher doses of thalidomide [104]. Several years later, however, a randomized, open label trial of docetaxel with or without thalidomide produced the encouraging result of a significant improvement in overall survival (25.9 months for thalidomide vs. 14.7 months for placebo, p = 0.0407) [105]. Venous thromboembolism was a significant adverse reaction to this regimen, which was effectively prevented later in the trial by the institution of low-molecular weight heparin for all participants. Thalidomide has also undergone phase II testing in combination with docetaxel and estramustine, which achieved a >50 % PSA response in 90 % of subjects and a PFS of 7.2 months [106].
Table 15.2
Phase II and III trials of thalidomide and lenalidomide for prostate cancer with reported results
Study title | Phase | N | Primary end point and results | Year | Reference |
|---|---|---|---|---|---|
Thalidomide | |||||
A randomized phase II trial of thalidomide, an angiogenesis inhibitor, in patients with androgen-independent prostate cancer | II | 63 | Primary end point not specified 0 % had an objective partial response, 14 % had PSA decline of >50 % | 2001 | Figg et al. [104] |
Randomized phase II trial of docetaxel plus thalidomide in androgen-independent prostate cancer | II | 75 | Overall survival and progression-free survival Median progression-free survival was 3.7 months in the docetaxel group and 5.9 months in the docetaxel + thalidomide group (p = 0.32). 18-month overall survival was 42.9 % in the docetaxel group and 68.2 % in the docetaxel + thalidomide group (p = 0.11) | 2004 | Dahut et al. [105] NCT00020046 |
Preclinical and clinical evaluation of estramustine, docetaxel and thalidomide combination in androgen-independent prostate cancer | II | 20 | Progression-free survival, objective response, PSA response Progression-free survival was 7.2 months. 20 % had a partial radiographic response. 90 % had a PSA decline of >50 % | 2007 | Figg et al. [106] NCT00083005 |
A double-blind randomized crossover study of oral thalidomide versus placebo for androgen-dependent prostate cancer treated with intermittent androgen ablation | III | 159 | Biochemical progression-free survival Median time to PSA progression was 15 months in the thalidomide group compared to 9.6 months in the placebo group (p = 0.21) | 2009 | Figg et al. [107] NCT00004635 |
Phase II trial of bevacizumab, thalidomide, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer | II | 60 | PSA response 90 % had PSA decline of >50 % | 2010 | Ning et al. [98] NCT00091364 |
Lenalidomide | |||||
Sargramostim (GM-CSF) and lenalidomide in castration-resistant prostate cancer (CRPC): Results from a phase I–II clinical trial | I–II | 32 | Objective tumor response, PSA response, and safety 18 % objective response rate. 13 % with PSA decline of >50 %. 22 % experience grade 3–4 toxicity | 2013 | Garcia et al. [111] NCT00939510 |
A phase 3 study to evaluate the efficacy and safety of Docetaxel and Prednisone (DP) with or without Lenalidomide (LEN) in patients with castrate-resistant prostate cancer (CRPC): the MAINSAIL trial | III | 1,059 | Overall survival Median overall survival was 77 weeks in the docetaxel + prednisone + lenalidomide group, compared to median not reached in the docetaxel + prednisone + placebo arm (p = 0.0017) | 2011 | Petrylak et al. [112] NCT00988208 |
The only double-blinded, randomized data on thalidomide for prostate cancer comes from a phase III trial of recurrent, but not castrate-resistant, disease. This study randomized patients with recurrent prostate cancer to androgen deprivation therapy followed by thalidomide or placebo, with a primary end point of time to PSA progression; this process was repeated at first evidence of progression, to allow for two treatment-and-progression phases. Though a trend towards a longer progression-free interval was present for both treatment phases, the difference was statistically significant for only the second phase (17.1 months for thalidomide vs. 6.6 months for placebo, p = 0.0002) [107].
Thalidomide has also been studied as part of a potent combination with bevacizumab, prednisone, and docetaxel. This regimen achieved a 90 % rate of >50 % PSA reduction, though it was not suitable for further study due to bone marrow suppression [98]. Due to a better side effect profile, interest and research efforts have shifted towards the thalidomide derivative lenalidomide [108].
Lenalidomide
Lenalidomide has a more tolerable side effect profile than thalidomide. This became clear during development for myeloma treatment, and has been confirmed in phase I dose-escalation trials of prostate cancer patients [109]. Since then, research into this new member of the thalidomide family has grown, with several trials currently underway (Table 15.2).
An earlier phase II trial suggested that a combination regimen of thalidomide, bevacizumab, prednisone, and docetaxel was highly active but limited by toxicity [98]. To build on this result, and hopefully to reduce toxicity with the substitution of thalidomide for lenalidomide, a phase II trial of lenalidomide, bevacizumab, prednisone, and docetaxel has been undertaken [NCT00942578]. This trial is still ongoing, but preliminary results have reported response rates of 79.3 % by radiography and 86.7 % by PSA [110]. Another phase I–II trial combining GM-CSF with lenalidomide demonstrated a favorable toxicity profile but only modest anti-tumor activity, with 12.5 % of CRPC patients achieving a PSA response of >50 % and 18 % of those with measurable disease having a radiographic response [111].
To try to replicate the potential favorable results observed with thalidomide plus docetaxel, a phase III trial was undertaken to test lenalidomide in combination with docetaxel for the treatment of CRPC. This multicenter, randomized, double-blinded trial, known as the MAINSAIL trial, enrolled a total of 1,059 patients. Disappointingly, the MAINSAIL trial was terminated early due to lack of efficacy, as lenalidomide failed to improve the primary outcome of overall survival [112].
Despite this significant setback, evaluation of other potential uses for lenalidomide in CRPC is ongoing. Lenalidomide is currently being studied in combination with paclitaxel [NCT00933426] as well as cyclophosphamide [NCT01093183] for the treatment of this disease.
Anti-angiogenic Tyrosine Kinase Inhibitors
Sunitinib
Many of the small-molecule TKIs act upon angiogenic pathways. They have the theoretical potential for a greater anti-angiogenic activity than single-target agents such as bevacizumab, as they often block multiple signaling pathways that contribute to angiogenesis. This fact also makes a broader range of side effects more likely, however. TKIs target tumor cells as well as the endothelial cells, turning off the autocrine and paracrine signals that promote tumor neovascularization [31].
Sunitinb malate inhibits a number of pro-angiogenic targets, including the VEGF receptors, PDGF receptors, the tyrosine-protein kinase KIT, colony stimulating factor 1 receptor, and receptor tyrosine kinases encoded by c-RET [31]. With FDA approval for the treatment of renal cell cancinoma and pancreatic neuroendocrine tumors, sunitinib is also one of the best-studied TKIs for the treatment of CRPC.
Anti-prostate cancer activity of sunitinib has been observed in several phase II trials (Table 15.3). The first phase II results for CRPC, reported in 2009, showed few PSA responses (2 of 34 patients, the primary end point), though it was noticed that radiographic responses were often discordant with PSA responses, raising the possibility that sunitinib’s anti-tumor activity was not being adequately captured by the PSA endpoint [113]. A similar trial of sunitinib monotherapy for CRPC after failure of cytotoxic chemotherapy also showed a low rate of >50 % PSA responses (12.1 %), though large numbers of patients saw smaller declines by both PSA and radiographic criteria [114]. Additional encouraging results were seen in combination with docetaxel and prednisone for chemotherapy-naïve CRPC, with a PSA response rate of 56.4 % and 12.6-month average PFS [115].
Table 15.3
Phase II and III, and selected phase I trials of TKIs for prostate cancer with reported results
Study title | Phase | N | Primary end point and results | Year | Reference |
|---|---|---|---|---|---|
Sunitinib | |||||
Phase II study of sunitinib in men with advanced prostate cancer | II | 34 | PSA response 6 % had PSA response >50 % regardless of docetaxel-naïve status | 2009 | Dror Michaelson et al. [113] NCT00299741 |
Sunitinib malate for metastatic castration-resistant prostate cancer following docetaxel-based chemotherapy | II | 36 | Progression-free survival Median progression-free survival was 19.4 weeks | 2010 | Sonpavde et al. [114] |
Sunitinib in combination with docetaxel and prednisone in chemotherapy-naive patients with metastatic, castration-resistant prostate cancer: a phase 1/2 clinical trial | I–II | 55 | PSA response 56 % had PSA decline of >50 % | 2012 | Zurita et al. [115] NCT00137436 |
A multicenter, randomized, double-blind, phase 3 study of sunitinib plus prednisone versus prednisone in patients with progressive metastatic castration-resistant prostate cancer after failure of a docetaxel-based chemotherapy regimen | III | 873 | Overall survival Trial was stopped early for futility. At the time of discontinuation, disease progression rates were 28.1 % in the sunitinib group and 47.3 % in the placebo group | 2010 | Ou et al. [116] NCT00676650 |
Sofafenib | |||||
A clinical phase II study with sorafenib in patients with progressive hormone-refractory prostate cancer: a study of the CESAR Central European Society for Anticancer Drug Research-EWIV | II | 47 | 12-week progression-free survival Median progression-free survival was 8 weeks | 2007 | Steinbild et al. [118] |
A phase II study of BAY 43-9006 (Sorafenib) in metastatic, androgen-independent prostate cancer | II | 22 | Progression (radiographic, biochemical, or symptomatic) Median progression-free survival was 1.8 months. No responses were seen by radiographic or biochemical measures | 2008 | Dahut et al. [119] NCT00090545 |
Final analysis of a phase II trial using sorafenib for metastatic castration-resistant prostate cancer | II | 24 | Progression-free survival Median progression-free survival was 3.7 months and median overall survival was 18.0 months | 2009 | Aragon-Ching et al. [120] NCT00090545 |
A phase II study of sorafenib in patients with chemo-naive castration-resistant prostate cancer | II | 28 | PSA response 3.6 % had PSA decline of >50 % | 2008 | Chi et al. [121] |
A phase II study of sorafenib in combination with bicalutamide in patients with chemotherapy-naive castration-resistant prostate cancer | II | 39 | PSA response 32 % had PSA decline of >50 % | 2012 | Beardsley et al. [122] |
Cabozantinib | |||||
Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial | II | 171 | Objective response rate and progression-free survival Median progression-free survival was 23.9 weeks with cabozantinib compared to 5.9 weeks in the placebo group (p < 0.001) | 2013 | Smith et al. [123] NCT00940225 |
Vandetanib | |||||
A phase II, double-blind, placebo-controlled, randomized study to assess the efficacy and safety of docetaxel (taxotere)/prednisolone/ZD6474 vs docetaxel/prednisolone/placebo in patients with hormone-refractory prostrate cancer (HRPC) | II | 86 | PSA response 40 % of subjects had a >50 % decline in PSA in the docetaxel + prednisolone + vandetanib group, compared to 67 % in the docetaxel + prednisolone group | 2011 | NCT00498797 |
Efficacy and Safety of Zactima™ in patients with castration-refractory metastatic prostate cancer | II | 110 | PSA progression-free survival at 4 months PSA progression-free survival was 18 % in the bicalutamide + vadetanib group and 16 % in the bicalutamide + placebo group | 2011 | NCT00659438 |
Pazopanib | |||||
Pazopanib hydrochloride after leuprolide acetate or goserelin acetate in treating patients with relapsed prostate cancer | II | 37 | Time to PSA progression Trial was stopped early due to high drop-out rates | 2012 | Ward et al. [128] NCT00454571 |
Cediranib | |||||
Phase I dose-escalation and pharmacokinetic study of AZD2171, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinase, in patients with hormone-refractory prostate cancer (HRPC) | I | 26 | Maximum tolerated dose Maximum tolerated dose was found to be 20 mg per day. PSA reductions were seen in 15 % of patients | 2007 | Ryan et al. [130] |
Based on these results, a placebo-controlled phase III trial of sunitinib in combination with prednisone for CRPC previously treated with docetaxel was undertaken [NCT00676650]. Unfortunately, this trial was stopped prematurely based on preliminary results indicating that the primary endpoint of overall survival would not show a benefit. Similar to the CALBG 90401 trial of bevacizumab for CRPC [95], sunitinib showed a significant improvement in PFS (5.6 vs. 3.7 months, p = 0.0077) but no improvement in overall survival (13.1 vs. 12.8 months, p = 0.58) [116].
Despite this setback, sunitinib is still undergoing active study for the management of prostate cancer. A single-arm phase II trial is examining sunitinib in the non-CRPC setting, as part of combined treatment with docetaxel and salvage radiation therapy for PSA recurrence after prostatectomy [NCT00734851. Sunitinib is also being tried in the neoadjuvant setting before prostatectomy [NCT00329043]. A randomized phase II is comparing sunitinib vs. dasatinib, plus abiraterone and prednisone, for control of CRPC [NCT01254864].
Sorafenib
Sorafenib is another small-molecule, multityrosine kinase inhibitor with a broad spectrum of activity. Anti-angiogenic activity is thought to derive from its inhibition of the VEGF receptors 2 and 3 and PDGF receptor β, as well as p38, c-kit, b-Raf, and c-Raf. By these pathways, sorafenib also exhibits direct anti-tumor and pro-apoptotic as well as anti-angiogenic effects [117].
Beginning in 2007, several phase II trials have evaluated sorafenib for CRPC (Table 15.3). When used as monotherapy for CRPC, the best response by RECIST was stable disease in 4 of 55 patients; two patients were responders by PSA, and 31 % overall met the primary endpoint of PFS at 12 weeks [118]. Another trial of sorafenib monotherapy for CRPC noticed a discordance between radiographic and PSA response; while most patients (21 of 22) progressed, 13 of 21 progressed only by PSA in the absence of any radiographic progression. Additionally, two patients initially experienced a remarkable reduction in bony metastatic disease [119]. After these encouraging results, patient accrual was continued; final results later reported 1 partial response and 11 with stable disease out of 24 total patients [120]. A separate trial of sorafenib monotherapy for CRPC also noticed a poor PSA response rate, with only 1 of 28 patients having a >50 % decline; however, in this trial most of the patients showed radiographic progression as well [121]. Sunitinib has been tested in combination with androgen deprivation therapy (bicalutamide) for CRPC with better results—nearly half (18 of 39) of patients achieved the primary outcome of either a PSA response or stable disease at >6 months [122]. As many of these patients had previously progressed on anti-androgen therapy, including bicalutamide, these results raise the possibility of synergistic effect of anti-androgen and anti-angiogenic therapy. However, this possibility has not yet undergone further testing, and there are not currently any ongoing trials of sorafenib in prostate cancer.
Cabozantinib
Cabozantinib is a small-molecule TKI that exerts anti-angiogenic action via inhibition of the VEGF receptor 2 and MET (the HGF receptor), a promoter of tumor invasion and metastasis [31]. It has additional anti-tumor effects via inhibition of c-ret and c-kit. Results from several ongoing trials of this agent are highly anticipated, after a remarkable response rate seen in early phase II data (Table 15.3). Cabozantinib has already been FDA-approved for the treatment of medullary thyroid cancer.
To date, only one phase II trial of cabozantinib for CRPC has been reported. In a randomized discontinuation design, 171 patients with CRPC were treated with cabozantinib monotherapy for a 12-week lead-in phase, during which 72 % of the 154 patients with measurable disease saw regression of soft tissue lesions; those 31 patients who had stable disease after the lead-in phase were randomly assigned to continue cabozantinib or to placebo. The trial was stopped early due to benefit; PFS during the post-randomization period was 23.9 weeks for cabozantinib vs. 5.9 weeks for placebo (p < 0.001). Additionally, significant reductions were seen in the secondary outcomes of bone pain and narcotic use [123].
After preliminary data from this trial were reported at the American Society of Clinical Oncology meeting in 2011, multiple trials have begun to expand upon these exciting results. The COMET-I trial, a phase III, double-blinded, placebo-controlled trial comparing cabozantinib to prednisone in the setting of previously treated CRPC is ongoing [NCT01605227]; COMET-II, comparing cabozantinib to combined mitoxantrone and prednisone for previously treated CRPC with the primary endpoint of pain response is currently recruiting patients [NCT01522443]. Additional phase II trials are ongoing as well, evaluating cabozantinib monotherapy in CRPC with visceral [NCT01834651] and bony [NCT01428219] metastasis, in non-metastatic disease [NCT01703065], in combination with abiraterone [NCT01995058], and also as first-line treatment in combination with anti-androgen therapy for metastatic but castrate-naïve prostate cancer [NCT01630590].
Vandetanib
Vandetanib is a small-molecule TKI that has been employed in the treatment of medullary thyroid carcinoma, where its activity is thought to derive from its inhibition of the RET oncogene. It also has anti-angiogenic activity via inhibition of the VEGF receptors 2 and 3, as well as the epidermal growth factor (EGF) receptor [124]. Vandetanib has also been studied in other malignancies, with a phase III trial for advanced non-small cell lung cancer showing a modest benefit in PFS (4.0 vs. 3.2 months for placebo, p < 0.0001) but no benefit OS [125].
The first data for vandetanib in prostate cancer came in 2007; a randomized, double-blinded phase II trial tested vandetanib in combination with docetaxel and prednisolone for chemotherapy-naïve CRPC (Table 15.3). Forty three patients were enrolled into each group; more patients in the vandetanib group withdrew (38) compared to placebo (29), which was driven by a higher rate of adverse events. The primary end point of PSA response was more common in the placebo group (29 patients vs. 17 patients for vandetanib) [NCT00498797]. Vandetanib was also tried in combination with bicalutamide for CRPC, without improvement in the primary endpoint of PSA progression, or in PFS or OS [NCT00659438]. There are no ongoing clinical trials of vandetanib for prostate cancer.
Pazopanib
This TKI is active on several angiogenic targets, including VEGF receptors 1–3 signaling, PDGF receptors α and β, and the FGF receptor [126]. It has been approved for use in advanced soft tissue sarcoma and in advanced renal cell carcinoma after phase III results showed significant improvement in PFS [127].
Several early trials of pazopanib in prostate cancer met significant hurdles (Table 15.3). One phase II trial of pazopanib for castrate-sensitive prostate cancer was terminated due to high drop-out rates, driven by toxicity in the pazopanib arm and protocol non-compliance in the placebo arm [128]. Another phase II trial of pazopanib in CRPC was terminated due to slow enrollment [NCT00945477]. Despite these setbacks, research in this area is ongoing. A randomized, double-blinded phase I/II combining pazopanib with docetaxel and prednisone in the setting of CRPC with unfavorable risk factors is currently recruiting [NCT01385228]. A phase II of neoadjuvent pazopanib before prostatectomy with a primary endpoint of risk of metastasis is planned but has not yet opened for recruitment [NCT01832259].
Cediranib
Cediranib has a large degree of overlap in its inhibitory spectrum with pazopanib, targeting the VEGF receptors 1–3, PDGF receptors α and β, and the FGF receptor, as well as c-KIT [129].
Early dose-escalation studies in prostate cancer reported 5 PSA reductions out of 26 patients, noting that these responses were often delayed, occurring after drug withdrawal (Table 15.3) [130]. Phase II data is still being awaited. A randomized phase II of docetaxel and prednisone with or without cediranib for CRPC is ongoing [NCT00527124]. Cediranib is also undergoing evaluation as monotherapy in CRPC previously treated with docetaxel [NCT00436956].
Other Classes and Novel Agents
Tasquinimod
Tasquinimod is a quinoline-3-carboxamide derivative which has been found to have anti-angiogenic effects in prostate cancer model systems. Its mechanism of action, elucidated using an in vitro prostate cancer model appears to be upregulation of thrombospondin-1, an inhibitor of HIF-1, resulting in a down-regulation of VEGF production [131]. This novel mechanism of action makes this a particularly interesting agent, especially as it may allow for combination therapy with other drugs targeting the VEGF pathway at multiple points.
A single phase II clinical trial has been reported, and its results were encouraging (Table 15.4). This relatively large (201 patients), randomized, double-blinded study of tasquinimod for chemotherapy-naïve, metastatic CRPC found a significant improvement in PFS (7.6 vs. 3.3 months for placebo, p = 0.0042) [132]. Updated results from this trial have continued to show a PFS advantage to tasquinimod, as well as a non-statistically significant trend towards longer OS [133].
Table 15.4
Phase II and III trials of novel angiogenesis inhibitors for prostate cancer
Study title | Phase | N | Primary end point and results | Year | Reference |
|---|---|---|---|---|---|
Everolimus | |||||
RAD001 in patients with metastatic, hormone-refractory prostate cancer | II | 35 | PSA response 0 % had PSA decline >50 % | 2013 | NCT00629525 |
Phase 2 trial of single-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08) | II | 37 | Progression-free survival Progression-free survival was 35 % at 12 weeks | 2013 | Templeton et al. [135] NCT00976755 |
The use of RAD001 with docetaxel in the treatment of metastatic, androgen-independent prostate cancer | I–II | No study results reported | 2013 | NCT00459186 | |
Tasquinimod | |||||
Phase II randomized double blind placebo-controlled study to determine the efficacy of ABR-215050 in asymptomatic patients with metastatic castrate-resistant prostate cancer | II | 201 | Progression-free survival at 6 months 69 % of subjects receiving tasquinimod were progression-free at 6 months, compared to 37 % in the placebo group (p < 0.001) | 2011 | Pili et al. [132] NCT00560482
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|