β-Thalassemia is a genetic disorder caused by mutations in the β-globin gene and characterized by chronic anemia caused by ineffective erythropoiesis, and accompanied by a variety of serious secondary complications such as extramedullary hematopoiesis, splenomegaly, and iron overload. In the past few years, numerous studies have shown that such secondary disease conditions have a genetic basis caused by the abnormal expression of genes with a role in controlling erythropoiesis and iron metabolism. In this article, the most recent discoveries related to the mechanism(s) responsible for anemia/ineffective erythropoiesis and iron overload are discussed in detail. Particular attention is paid to the pathway(s) controlling the expression of hepcidin, which is the main regulator of iron metabolism, and the Epo/EpoR/Jak2/Stat5 signaling pathway, which regulates erythropoiesis. Better understanding of how these pathways function and are altered in β-thalassemia has revealed several possibilities for development of new therapeutic approaches to treat of the complications of this disease.
β-Thalassemia
Genetic Causes, Consequences and Pleiotropic Effects
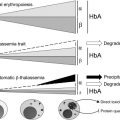
As discussed in more detail in the overview by Sankaran and Nathan elsewhere in this issue, β-thalassemia is an inherited disorder characterized by mutations in the gene encoding β-globin that lead to the quantitative reduction or, in the most severe cases, the total absence of β-globin synthesis in human erythroid cells. As a consequence, α-globin chains accumulate in excess, forming aggregates that impair erythroid cell maturation, which ultimately leads to a chronic hemolytic anemia and ineffective erythropoiesis (IE) ( Fig. 1 ). The severity of the clinical manifestations in β-thalassemia varies widely, ranging from patients that are almost asymptomatic to individuals who suffer from severe anemia and require regular blood transfusion to sustain life. In general, the clinical severity of the disease correlates with the size of the free α-chain pool and the degree of imbalance between the production of α- and β-like globin chains.
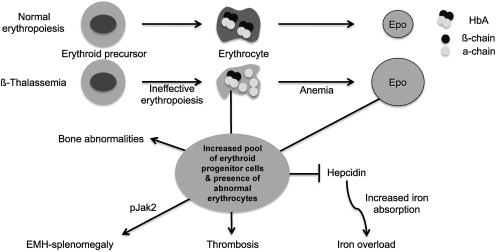
The α/β globin chain imbalance is responsible for the hemolysis of red blood cells (RBCs) and for the premature death (apoptosis) of erythroid precursors in the bone marrow and at extramedullary sites (see Fig. 1 ). The α-globin chain aggregates form inclusion bodies responsible for oxidative stress and membrane damage within RBCs and immature developing erythroblasts. These events are followed by the premature death of many late erythroid precursors in the bone marrow and spleen. Together, these phenomena are designated as IE. The anemia and resulting hypoxia lead to a dramatic increase in serum erythropoietin (Epo) levels in an attempt to compensate for the reduced oxygen-carrying capacity. The marked increase in Epo stimulation, if it is not inhibited by proper transfusion therapy, can lead to uncontrolled expansion of erythroid precursors in the marrow as well as in other sites, such as the spleen and liver, leading to extramedullary hematopoiesis (EMH) (see Fig. 1 ).
The defect in hemoglobin synthesis that occurs in β-thalassemia leads to the development of pleiotropic effects on different body compartments. For example, one of the consequences of IE and EMH is splenomegaly (see Fig. 1 ). The abnormal and damaged RBCs that are produced in β-thalassemia are sequestered by the reticuloendothelial system in the spleen, causing its enlargement. This enlargement leads to increased sequestration of RBCs, including transfused RBCs, in the spleen, with worsening of the anemia and an increase in transfusion requirement. Increased Epo synthesis is also reflected in marrow expansion, leading to bone marrow hyperplasia, bone deformities, and osteopenia (see Fig. 1 ), which contribute to increased morbidity as the disease progresses.
However, iron overload is the principal and multifaceted complication of β-thalassemia. Physiologically, it is caused by an increased absorption of iron from the gastrointestinal (GI) tract as a consequence of IE, and is greatly aggravated by chronic transfusion therapy. Thus, transfusion-independent individuals with thalassemia intermedia have a slower progression of iron overload and generally develop complications later in life compared with patients with thalassemia major who are chronically transfused (see Fig. 1 ). A remarkable variability of tissue iron distribution has been observed in β-thalassemia; liver, heart, and endocrine glands are the organs most severely affected. Accumulation of iron at these sites (siderosis) leads to oxidative damage as a result of the generation of reactive oxygen species (ROS). Death from cardiac failure is the most common clinical consequence of iron excess. Pathologic analysis of cardiac tissue has shown that this occurs initially by hypertrophy and dilatation followed by degeneration of myocardial fibers and fibrosis. Endocrine problems are also related to iron overload (see Fig. 1 ). These problems include hypogonadism, which leads to disturbances of growth and sexual maturation, as well as hypothyroidism, hypoparathyroidism, and diabetes mellitus, which are seen in variable numbers of patients. Therefore, effective iron chelation therapy is a critical component in the management of thalassemia. For more than 3 decades, deferoxamine (Desferal) was the chelator of choice. Although this drug is capable of placing all patients into net negative iron balance, it has a disadvantage because it must be infused subcutaneously for 8 to 12 hours, 5 to 7 days a week for the duration of life. Not surprisingly, adherence to such a regimen is generally poor. More recently, 2 orally effective iron chelators, Exjade (deferasirox) and deferiprone (Ferriprox), have been developed to overcome this limitation. Iron overload and chelation therapy are discussed in greater detail elsewhere in this issue in the article by Porter and Shah.
The only definitive cure for thalassemia is the transplant of hematopoietic stem cells (HSC) from cord blood or the bone marrow. This topic is discussed in detail elsewhere in this issue in the article by Gaziev and Lucarelli, as well as that by Kanathezhath and Walters. Splenectomy is another facet in the management of β-thalassemia. However, removal of the spleen can lead to further problems such as an increased risk of infections, pulmonary hypertension, and thrombosis, and is usually considered only in the most severe cases of splenomegaly. New approaches to treat/cure the disease are being developed, such as gene therapy, discussed in the article by Bank elsewhere in this issue, or the use of induced pluripotent stem cells as alternatives to the use of HSC.
Mouse Models of β-Thalassemia
The use of mouse models of β-thalassemia has been of fundamental importance in clarifying the molecular mechanisms responsible for disease. The 2 available mouse models of β-thalassemia intermedia are th1 / th1 and th3 /+. In the th1 / th1 mouse, the deletion removes the β major gene in the homozygous state, producing hemoglobin levels in the range of 9 to 10 g/dL. In the th3 /+ mouse model, the deletion removes both the β minor and β major genes in the heterozygous state. These mice have hemoglobin levels in the range of 8 to 9 g/dL. Both models have a degree of disease severity (hepatosplenomegaly, anemia, aberrant erythrocyte morphology) comparable with that of patients affected by transfusion-independent β-thalassemia intermedia. Their IE is characterized by a modest reduction in RBCs and an increase in reticulocytes. Homozygous ( th3 / th3 ) mice that completely lack any adult β-globin chain synthesis die late in gestation, precluding their use as a model for β-thalassemia major. To overcome this limitation, th3 / th3 mice are generated by transplantation of hematopoietic fetal liver cells (HFLCs) into lethally irradiated syngeneic adult recipients, the HFLCs being harvested from th3 / th3 embryos at 14.5 days of gestation. These mice exhibit severe anemia (3–4 g/dL) 6 to 8 weeks after transplant as well as low RBC levels and reticulocyte counts, together with massive splenomegaly and extensive EMH.
IE
The Epo/EpoR/Jak2/Stat5 Pathway and its Potential Effect(s) on Iron Intake in Erythroid Cells
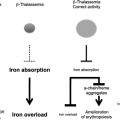
The hallmark of β-thalassemia is IE that stems from a lack of or reduced synthesis of β-globin, which leads to an excess of α-globin chains that aggregate and precipitate, adhering to the membrane of erythroid precursors. These α-globin aggregates cause cellular and membrane damage, apoptosis of the erythroid precursors in the bone marrow and generation of mature red cells that are abnormal and accumulate in limited numbers. Moreover, the production of red cells, EMH, and the anemia can change significantly over time. Therefore, β-thalassemia serves as a good example of the dynamic balance between expansion of the erythroid pool and production of red cells, and of the many factors that can alter this relationship with critical effects. When pathologic levels of damaged erythrocytes are trapped in the spleen they cause splenomegaly, anemia, and hypoxia. Anemia and hypoxia, in turn, stimulate Epo synthesis, which increases the number of erythroid precursors and abnormal mature red cells in circulation. This situation exacerbates the trapping of erythrocytes in the spleen, thereby worsening the splenomegaly. Moreover, increased erythropoiesis augments iron absorption. Iron overload can increase the formation of ROS, causing damage to many organs and further aggravating the anemia. Blood transfusion is an effective method for ameliorating the anemia and the consequences of IE. But good adherence to iron chelation therapy is necessary to prevent the detrimental effects of iron overload.
Epo is a 34-kDa renal glycoprotein that functions as the main regulator of erythropoiesis, both under basal and stress conditions. Epo binds to its specific receptor, the erythropoietin receptor (EpoR), at the surface of erythroid cells. EpoR is expressed by the earliest erythroid progenitors at the burst-forming unit-erythroid (BFU-E) stage. The Epo/EpoR signaling pathway begins with dimerization of the receptor and activation of the tyrosine-Janus kinase 2 (Jak2), which preassembles at a conserved site in the cytoplasmic domain of the EpoR. Jak2 catalyzes transfer of the γ-phosphate group of ATP to the hydroxyl groups of specific tyrosine residues in signal transduction molecules and mediates signaling downstream of cytokine receptors after ligand-induced autophosphorylation of itself and of tyrosine residues of the receptor. In particular, Jak2 mediates the phosphorylation of tyrosine residues localized in a conserved cytoplasmic domain of the EpoR. The resulting phosphorylated tyrosine residues of the EpoR function as a scaffold or docking sites for the assembly of signal transduction factors containing Src-homology 2 (SH2) domains. The main downstream effectors of Jak2 are a family of transcription factors known as signal transducers and activators of transcription (Stat). In erythroid cells, the main target is Stat5, which, on phosphorylation, translocates to the nucleus as a dimer to drive expression of target genes. The ultimate effect of Jak2 and Stat5 activation is to induce multiple signaling pathways designed to regulate erythroid proliferation and differentiation, and to protect the cells from apoptosis.
Polycythemia vera (PV), essential thombocytosis, and primary myelofibrosis are classified as myeloproliferative disorders, a subgroup of myeloid malignancies. These are clonal stem cell diseases characterized by an expansion of morphologically mature cells of the granulocyte, erythroid, megakaryocyte, or monocyte lineage. Several studies have described a close association between an activating JAK2 mutation (Val 617 to Phe; JAK2V617F) and these disorders. This mutation is believed to prevent the pseudokinase domain from inhibiting the kinase domain, resulting in a constitutively active state of the protein. Thus, JAK2V617F confers constitutive kinase activation. In erythroid cells this leads to STAT5 phosphorylation and Epo-independent erythroid colony formation.
Mice lacking Stat5 expression (Stat5 –/– ) have shown early lethality associated with microcytic anemia and enhanced apoptosis of early erythroblasts. Although the anemia in these mice correlates with loss of expression of the antiapoptotic Bcl-X L gene and enhanced apoptosis, additional analyses indicate that Stat5 –/– mice have a significant decrease in expression of the iron transporter transferrin receptor-1 (Tfr1). Therefore, it is possible that erythroid cells might express higher levels of Tfr1 under conditions of constitutive activation or upregulation of Jak2. Potential consequences of Jak2 modulation of iron intake in β-thalassemic red cells are discussed in the next section.
New Studies on Jak2 and IE in β-Thalassemia
Original ferrokinetic studies and analysis of erythroid precursors in β-thalassemia indicated that many of these cells die in the marrow and extramedullary sites, and suggested that the relative excess of α chains triggering apoptosis was responsible for IE. However, several recent observations suggest that the balance between proliferation and differentiation in some of the erythroid precursors might be different under normal conditions and those of IE. These results and those of new studies in mice (discussed later) suggest that the mechanism(s) and various factors associated with the process known as IE have not yet been completely elucidated.
The splenomegaly exhibited by th3 /+ mice is similar to that observed in patients affected by β-thalassemia intermedia (transfusion independent). Studies on th3 /+ mice have shown that their spleen fills with erythroid precursors and sequesters abnormal/damaged erythrocytes, likely contributing to lowering of the hemoglobin level over time. Similar observations have been confirmed in splenic specimens from patients with thalassemia intermedia. However, analysis of the erythroid precursors in both the bone marrow and spleen in these animals indicated that a large number of these cells were actively proliferating, whereas the proportion of cells undergoing apoptosis, although higher than in normal mice, was modest. Compared with wild-type animals, th3 /+ mice have a higher number of erythroid cells associated with the expression of cell cycle-promoting genes such as EpoR , Jak2 (see Fig. 1 ), Cyclin-A , Cdk2 , and Ki-67 , together with increased levels of the Bcl-X L protein. These cells also differentiate less than the corresponding normal cells in vitro. Based on this observation, we can speculate that thalassemic patients may increase the number of erythroid precursors in their spleen and liver over time, this being one factor leading to hepatosplenomegaly. In turn, this factor also exacerbates the anemia in β-thalassemia.
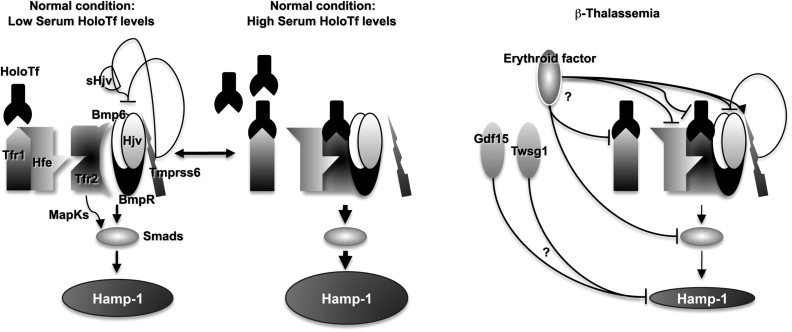
The increase of Epo levels in response to anemia and hypoxia has already been discussed. Based on the observations made earlier, it is reasonable to expect that increased Epo levels could also activate the EpoR/Jak2 pathway, leading to a physiologic gain of function of Jak2. Under these circumstances, the persistent phosphorylation of Jak2 might lead to an increased number of erythroid progenitor cells. Therefore, suppression of Jak2 activity might modulate IE. Based on this model, we showed that use of a Jak2 inhibitor has a beneficial effect in limiting IE, splenomegaly, and the number of erythroid progenitor cells in β-thalassemia. In particular, limiting the number of erythroid progenitors might have a beneficial effect on iron metabolism, because it has been suggested that these cells are the most plausible source of the erythroid factor, whose function is to increase iron absorption by repressing hepcidin expression, as shown in Figs. 1 and 2 and further discussed later. Therefore, future studies need to address whether the use of Jak2 inhibitors could also limit iron absorption in β-thalassemia.
Activation of the Epo/EpoR/Jak2 pathway is not likely to be the only cause for the limited erythroid differentiation observed in β-thalassemia. For example, in the absence of other erythroid defects, mutations responsible for the constitutive activation of Jak2 lead to the development of PV rather than IE. Therefore, it is possible to predict that other factors and/or abnormal physiologic conditions present in β-thalassemia interfere with erythroid cell differentiation. Among the possible factors acting together with Jak2, iron overload, ROS, the unbalanced synthesis of globin chains and/or heme can be considered. Iron is essential for all cells but is toxic in excess. We recently focused on the potential role of iron and heme in modulating IE. Our hypothesis was that thalassemic erythroid cells accumulate an excess of toxic heme associated with free α chains, leading to the formation of ROS, which has been associated with red cell hemolysis and altered differentiation. Our preliminary data suggest that this is the case and that if the hemoglobin content per cell (mean corpuscular hemoglobin [MCH]) and the overall amount of heme are reduced in thalassemic erythroid cells, both ROS and the accumulation of α-chains on red cell plasma membranes are reduced, ameliorating not only the quality and life span of the RBCs but also the anemia and IE (Gardenghi and colleagues, unpublished data, 2010). The amelioration of IE was associated with decreased splenomegaly and a better balance between erythroid precursors and mature RBCs. Based on these preliminary observations, we speculate that ROS play a role in modulating the balance between proliferation and differentiation of the erythroid cells. Additional studies point to the role of ROS in cell differentiation in erythropoiesis, stem cells, and cancer, supporting this hypothesis.
Serum iron is bound to transferrin and enters erythroid cells primarily via receptor-mediated endocytosis of the transferrin/Tfr1 complex. Tfr1 is essential for developing erythrocytes. Reduced Tfr1 expression is associated with anemia. Stat5-null mice are severely anemic and die perinatally. Two studies have associated Stat5 with iron homeostasis, showing that ablation of Stat5 leads to a dramatic reduction in both the mRNA and protein levels of iron regulatory protein 2 (IRP-2) and Tfr1 . Both genes have been shown to be direct transcriptional targets of Stat5, establishing a clear link between EpoR/Jak2/Stat5 signaling and iron metabolism. As we proposed previously, reduced iron intake in erythroid cells limits the formation of toxic α-chain/heme aggregates and ROS formation, having a beneficial effect on IE. This finding has been supported indirectly by the work of Ginzburg and colleagues. In their studies, iron delivery to erythroid cells in mice affected by β-thalassemia intermedia was modulated by administration of apo-transferrin. This finding was associated with reduction of splenomegaly and IE, improvement in hemoglobin levels, and an increased number of RBCs. The hemoglobin content per cell (MCH) was also decreased, as was the formation of membrane-bound α-chain aggregates. These data reinforce the notion that decreasing iron availability, either by decreasing iron absorption or transferrin saturation, is beneficial to abnormal erythroid cells. Therefore, based on the association between Jak2 and Tfr1, Jak2 inhibitors might also ameliorate IE by decreasing expression of Tfr1, iron intake, formation of toxic α-chain/heme aggregates, and ROS. In conclusion, use of Jak2 inhibitors in β-thalassemia might also be beneficial because of their role in controlling iron metabolism in erythroid cells.
IE
The Epo/EpoR/Jak2/Stat5 Pathway and its Potential Effect(s) on Iron Intake in Erythroid Cells
The hallmark of β-thalassemia is IE that stems from a lack of or reduced synthesis of β-globin, which leads to an excess of α-globin chains that aggregate and precipitate, adhering to the membrane of erythroid precursors. These α-globin aggregates cause cellular and membrane damage, apoptosis of the erythroid precursors in the bone marrow and generation of mature red cells that are abnormal and accumulate in limited numbers. Moreover, the production of red cells, EMH, and the anemia can change significantly over time. Therefore, β-thalassemia serves as a good example of the dynamic balance between expansion of the erythroid pool and production of red cells, and of the many factors that can alter this relationship with critical effects. When pathologic levels of damaged erythrocytes are trapped in the spleen they cause splenomegaly, anemia, and hypoxia. Anemia and hypoxia, in turn, stimulate Epo synthesis, which increases the number of erythroid precursors and abnormal mature red cells in circulation. This situation exacerbates the trapping of erythrocytes in the spleen, thereby worsening the splenomegaly. Moreover, increased erythropoiesis augments iron absorption. Iron overload can increase the formation of ROS, causing damage to many organs and further aggravating the anemia. Blood transfusion is an effective method for ameliorating the anemia and the consequences of IE. But good adherence to iron chelation therapy is necessary to prevent the detrimental effects of iron overload.
Epo is a 34-kDa renal glycoprotein that functions as the main regulator of erythropoiesis, both under basal and stress conditions. Epo binds to its specific receptor, the erythropoietin receptor (EpoR), at the surface of erythroid cells. EpoR is expressed by the earliest erythroid progenitors at the burst-forming unit-erythroid (BFU-E) stage. The Epo/EpoR signaling pathway begins with dimerization of the receptor and activation of the tyrosine-Janus kinase 2 (Jak2), which preassembles at a conserved site in the cytoplasmic domain of the EpoR. Jak2 catalyzes transfer of the γ-phosphate group of ATP to the hydroxyl groups of specific tyrosine residues in signal transduction molecules and mediates signaling downstream of cytokine receptors after ligand-induced autophosphorylation of itself and of tyrosine residues of the receptor. In particular, Jak2 mediates the phosphorylation of tyrosine residues localized in a conserved cytoplasmic domain of the EpoR. The resulting phosphorylated tyrosine residues of the EpoR function as a scaffold or docking sites for the assembly of signal transduction factors containing Src-homology 2 (SH2) domains. The main downstream effectors of Jak2 are a family of transcription factors known as signal transducers and activators of transcription (Stat). In erythroid cells, the main target is Stat5, which, on phosphorylation, translocates to the nucleus as a dimer to drive expression of target genes. The ultimate effect of Jak2 and Stat5 activation is to induce multiple signaling pathways designed to regulate erythroid proliferation and differentiation, and to protect the cells from apoptosis.
Polycythemia vera (PV), essential thombocytosis, and primary myelofibrosis are classified as myeloproliferative disorders, a subgroup of myeloid malignancies. These are clonal stem cell diseases characterized by an expansion of morphologically mature cells of the granulocyte, erythroid, megakaryocyte, or monocyte lineage. Several studies have described a close association between an activating JAK2 mutation (Val 617 to Phe; JAK2V617F) and these disorders. This mutation is believed to prevent the pseudokinase domain from inhibiting the kinase domain, resulting in a constitutively active state of the protein. Thus, JAK2V617F confers constitutive kinase activation. In erythroid cells this leads to STAT5 phosphorylation and Epo-independent erythroid colony formation.
Mice lacking Stat5 expression (Stat5 –/– ) have shown early lethality associated with microcytic anemia and enhanced apoptosis of early erythroblasts. Although the anemia in these mice correlates with loss of expression of the antiapoptotic Bcl-X L gene and enhanced apoptosis, additional analyses indicate that Stat5 –/– mice have a significant decrease in expression of the iron transporter transferrin receptor-1 (Tfr1). Therefore, it is possible that erythroid cells might express higher levels of Tfr1 under conditions of constitutive activation or upregulation of Jak2. Potential consequences of Jak2 modulation of iron intake in β-thalassemic red cells are discussed in the next section.
New Studies on Jak2 and IE in β-Thalassemia
Original ferrokinetic studies and analysis of erythroid precursors in β-thalassemia indicated that many of these cells die in the marrow and extramedullary sites, and suggested that the relative excess of α chains triggering apoptosis was responsible for IE. However, several recent observations suggest that the balance between proliferation and differentiation in some of the erythroid precursors might be different under normal conditions and those of IE. These results and those of new studies in mice (discussed later) suggest that the mechanism(s) and various factors associated with the process known as IE have not yet been completely elucidated.
The splenomegaly exhibited by th3 /+ mice is similar to that observed in patients affected by β-thalassemia intermedia (transfusion independent). Studies on th3 /+ mice have shown that their spleen fills with erythroid precursors and sequesters abnormal/damaged erythrocytes, likely contributing to lowering of the hemoglobin level over time. Similar observations have been confirmed in splenic specimens from patients with thalassemia intermedia. However, analysis of the erythroid precursors in both the bone marrow and spleen in these animals indicated that a large number of these cells were actively proliferating, whereas the proportion of cells undergoing apoptosis, although higher than in normal mice, was modest. Compared with wild-type animals, th3 /+ mice have a higher number of erythroid cells associated with the expression of cell cycle-promoting genes such as EpoR , Jak2 (see Fig. 1 ), Cyclin-A , Cdk2 , and Ki-67 , together with increased levels of the Bcl-X L protein. These cells also differentiate less than the corresponding normal cells in vitro. Based on this observation, we can speculate that thalassemic patients may increase the number of erythroid precursors in their spleen and liver over time, this being one factor leading to hepatosplenomegaly. In turn, this factor also exacerbates the anemia in β-thalassemia.
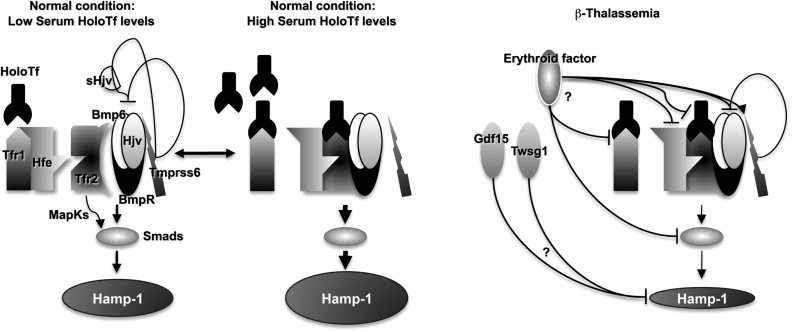
The increase of Epo levels in response to anemia and hypoxia has already been discussed. Based on the observations made earlier, it is reasonable to expect that increased Epo levels could also activate the EpoR/Jak2 pathway, leading to a physiologic gain of function of Jak2. Under these circumstances, the persistent phosphorylation of Jak2 might lead to an increased number of erythroid progenitor cells. Therefore, suppression of Jak2 activity might modulate IE. Based on this model, we showed that use of a Jak2 inhibitor has a beneficial effect in limiting IE, splenomegaly, and the number of erythroid progenitor cells in β-thalassemia. In particular, limiting the number of erythroid progenitors might have a beneficial effect on iron metabolism, because it has been suggested that these cells are the most plausible source of the erythroid factor, whose function is to increase iron absorption by repressing hepcidin expression, as shown in Figs. 1 and 2 and further discussed later. Therefore, future studies need to address whether the use of Jak2 inhibitors could also limit iron absorption in β-thalassemia.
Activation of the Epo/EpoR/Jak2 pathway is not likely to be the only cause for the limited erythroid differentiation observed in β-thalassemia. For example, in the absence of other erythroid defects, mutations responsible for the constitutive activation of Jak2 lead to the development of PV rather than IE. Therefore, it is possible to predict that other factors and/or abnormal physiologic conditions present in β-thalassemia interfere with erythroid cell differentiation. Among the possible factors acting together with Jak2, iron overload, ROS, the unbalanced synthesis of globin chains and/or heme can be considered. Iron is essential for all cells but is toxic in excess. We recently focused on the potential role of iron and heme in modulating IE. Our hypothesis was that thalassemic erythroid cells accumulate an excess of toxic heme associated with free α chains, leading to the formation of ROS, which has been associated with red cell hemolysis and altered differentiation. Our preliminary data suggest that this is the case and that if the hemoglobin content per cell (mean corpuscular hemoglobin [MCH]) and the overall amount of heme are reduced in thalassemic erythroid cells, both ROS and the accumulation of α-chains on red cell plasma membranes are reduced, ameliorating not only the quality and life span of the RBCs but also the anemia and IE (Gardenghi and colleagues, unpublished data, 2010). The amelioration of IE was associated with decreased splenomegaly and a better balance between erythroid precursors and mature RBCs. Based on these preliminary observations, we speculate that ROS play a role in modulating the balance between proliferation and differentiation of the erythroid cells. Additional studies point to the role of ROS in cell differentiation in erythropoiesis, stem cells, and cancer, supporting this hypothesis.
Serum iron is bound to transferrin and enters erythroid cells primarily via receptor-mediated endocytosis of the transferrin/Tfr1 complex. Tfr1 is essential for developing erythrocytes. Reduced Tfr1 expression is associated with anemia. Stat5-null mice are severely anemic and die perinatally. Two studies have associated Stat5 with iron homeostasis, showing that ablation of Stat5 leads to a dramatic reduction in both the mRNA and protein levels of iron regulatory protein 2 (IRP-2) and Tfr1 . Both genes have been shown to be direct transcriptional targets of Stat5, establishing a clear link between EpoR/Jak2/Stat5 signaling and iron metabolism. As we proposed previously, reduced iron intake in erythroid cells limits the formation of toxic α-chain/heme aggregates and ROS formation, having a beneficial effect on IE. This finding has been supported indirectly by the work of Ginzburg and colleagues. In their studies, iron delivery to erythroid cells in mice affected by β-thalassemia intermedia was modulated by administration of apo-transferrin. This finding was associated with reduction of splenomegaly and IE, improvement in hemoglobin levels, and an increased number of RBCs. The hemoglobin content per cell (MCH) was also decreased, as was the formation of membrane-bound α-chain aggregates. These data reinforce the notion that decreasing iron availability, either by decreasing iron absorption or transferrin saturation, is beneficial to abnormal erythroid cells. Therefore, based on the association between Jak2 and Tfr1, Jak2 inhibitors might also ameliorate IE by decreasing expression of Tfr1, iron intake, formation of toxic α-chain/heme aggregates, and ROS. In conclusion, use of Jak2 inhibitors in β-thalassemia might also be beneficial because of their role in controlling iron metabolism in erythroid cells.
Related posts:
 HbE/β-Thalassemia: Basis of Marked Clinical Diversity
HbE/β-Thalassemia: Basis of Marked Clinical Diversity
 Protein Quality Control During Erythropoiesis and Hemoglobin Synthesis
Protein Quality Control During Erythropoiesis and Hemoglobin Synthesis
 Iron Overload in Thal assemia and Related Conditions: Therapeutic Goals and Assessment of Response to Chelation Therapies
Iron Overload in Thal assemia and Related Conditions: Therapeutic Goals and Assessment of Response to Chelation Therapies
 Lower Gastrointestinal Tract Cancer Predisposition Syndromes
Lower Gastrointestinal Tract Cancer Predisposition Syndromes
 Anemia, Ineffective Erythropoiesis, and Hepcidin: Interacting Factors in Abnormal Iron Metabolism Leading to Iron Overload in β-Thalassemia
Hemoglobin Gene Therapy for β-Thalassemia
Anemia, Ineffective Erythropoiesis, and Hepcidin: Interacting Factors in Abnormal Iron Metabolism Leading to Iron Overload in β-Thalassemia
Hemoglobin Gene Therapy for β-Thalassemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


