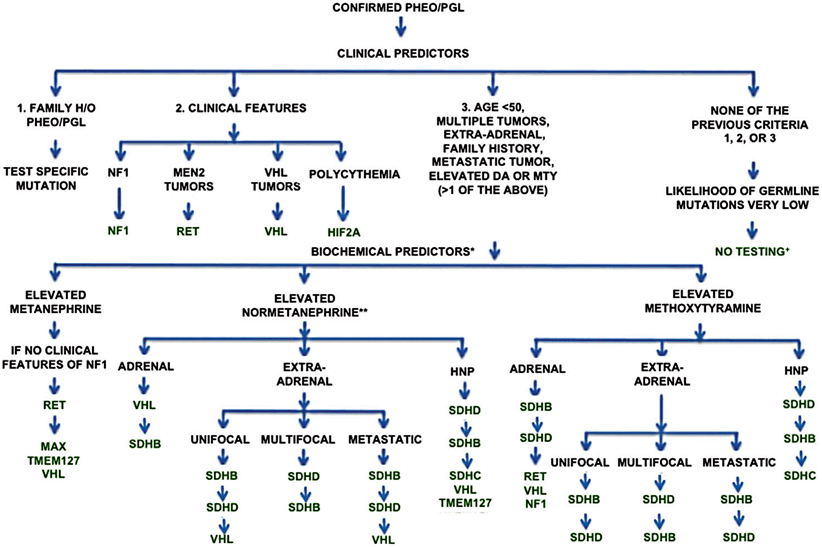
Fig. 1
A 31-year-old male noted to have a unilateral neck mass following an upper respiratory infection has a paraganglioma in the right carotid body ( white arrows). (Images courtesy of John York, M.D., Department of Radiology, Naval Medical Center Portsmouth, Virginia)
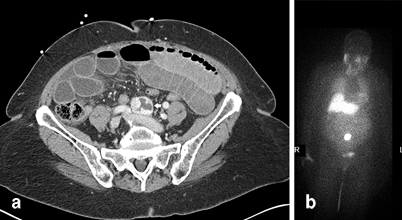
Fig. 2
An 83-year-old female, who presented with a small bowel obstruction, noted to have a mass at the aortic bifurcation found incidentally on CT. Subsequent planar and SPECT/ I-123-MIBG images demonstrated an organ of Zuckerkandl paraganglioma ( white arrows). CT computed tomography, SPECT single photon emission computed tomography, MIBG metaiodobenzylguanidine. (Images courtesy of Thomas Nelson, M.D., Department of General Surgery, Naval Hospital Camp Pendleton and Robert Marks, M.D., Department of Radiology, Naval Medical Center San Diego)
In the chromaffin cell , tyrosine is first converted to 3,4-dihydroxyphenylalanine (DOPA) by tyrosine hydroxylase. This is the rate-limiting step of catecholamine formation and serves as a point at which the physician can block catecholamine formation, either as part of the patient’s preoperative preparation or as palliative treatment for unresectable disease with metyrosine. DOPA is then converted to dopamine by aromatic l-amino acid decarboxylase. Dopamine is then converted to norepinephrine by dopamine β-hydroxylase and norepinephrine is finally converted to epinephrine by phenylethanolamine N-methyltransferase (PMNT). PMNT is almost exclusively located in the adrenal medulla. Further, cortisol acts as a necessary cofactor in PMNT’s mechanism of action. Thus, chromaffin tumors producing only norepinephrine as manifested by high normetanephrine levels are typically paraganglioma and not pheochromocytoma [2] . The exception to this rule is the adrenal pheochromocytomas found in von Hippel Lindau disease [11].
Presentation
Given its rarity, pheochromocytoma and paraganglioma are generally not routinely sought out as a cause of secondary hypertension . Further complicating the issue, as many as 30 % of patients with pheochromocytoma or paraganglioma are either normotensive or experience only orthostatic hypotension [13]. However, in cases of heightened clinical suspicion, such as an early onset hypertension, or in a patient who presents with episodic, paroxysmal hypertension accompanied by the classic constellation of symptoms including headaches, palpitations and diaphoresis, the diagnosis of pheochromocytoma and paraganglioma should be considered. Only 10 % of patients will present with the classic constellation of symptoms and the key to making the diagnosis is first considering the possibility. Other symptoms of catecholamine excess may include pallor, hyperglycemia, anxiety, or a sense of foreboding, nausea, and/or vomiting. If these symptoms accompany micturition, a paraganglioma near the bladder should be considered. Further, a patient who demonstrates an unusually high blood pressure in response to an invasive procedure (such as general anesthesia, surgery, or interventional radiological interrogation) should undergo a workup for pheochromocytoma. The diagnosis of pheochromocytoma should also be entertained whenever an adrenal mass is uncovered on abdominal imaging either as an adrenal incidentaloma or as part of a metastatic workup for a known malignancy. Pheochromocytomas account for approximately 5–7 % of adrenal incidentalomas [14] and perhaps 20–30 % of current pheochromocytomas are discovered first as adrenal incidentalomas [15]. Left untreated, pheochromocytomas can cause catecholamine-induced cardiomyopathy, congestive heart failure, myocardial infarction, stroke, and death.
Workup
Epinephrine and norepinephrine are degraded by methylation through catechol-O-methyltransferase to metanephrine and normetanephrine , respectively, or by deamination through monoamine oxidase to vanillymandelic acid [16]. Measurements of plasma and urine catecholamines are unhelpful because the half-lives of the catecholamines are extremely short. In contrast, the metanephrine by-products have a longer half-life. Plasma-free metanephrines have a high sensitivity at 96–100 % but a lower specificity of 85–89 % [11]. Twenty-four-hour urine fractionated metanephrines have a sensitivity of 77–90 %, but a high specificity of 93–98 % [11]. In general, plasma metanephrines are the screening test of choice due to the ease of obtaining samples and the higher sensitivity [17, 18]. Metanephrine levels greater than two to three times the upper limit of normal carry a near 100 % positive predictive value for a pheochromocytoma and functional paraganglioma . Similarly, a metanephrine level that is less than one time the upper limit of normal has a very high negative predictive value [2]. When the metanephrine levels are between one and two times the upper limit of normal, there is a 30 % chance of having a pheochromocytoma or functional paraganglioma [19]. Unfortunately, no clinical factors have been shown to distinguish between patients with and without pheochromocytoma or functional paraganglioma in this latter group of patients. Of note, paraganglioma typically lack the N-methyl transferase necessary to convert norepinephrine to epinephrine , so a paraganglioma may have normal levels of metanephrine and only elevated normetanephrines while the total metanephrine should still be elevated. When testing metanephrine levels, it is important to eliminate confounding factors: the patient should ideally be in a stress-free state, lie supine for 20 min prior to phlebotomy and should refrain from intake of caffeine or other stimulants prior to testing. The list of medications that can potentially affect biochemical testing includes tricyclic antidepressants, levodopa, over-the-counter (OTC) decongestants, amphetamines, buspirone and other antipsychotic agents, prochlorperazine, reserpine, cocaine, nicotine, and ethanol. Withdrawal from clonidine and other stimulants can also affect biochemical testing [2]. In tumors that can secrete dopamine such as those caused by SDHB (see section “Genetic Syndromes”) or SDHD mutations, consideration should be given to testing for dopamine and its metabolite methoxytyramine [20]. In general, testing for metanephrines should be done on two separate occasions to confirm the diagnosis.
After biochemical confirmation of the diagnosis, anatomic imaging (computed tomography (CT) scan or magnetic resonance imaging (MRI)) should be obtained followed by a functional imaging study (metaiodobenzylguanidine (MIBG) or positron emission tomography (PET) scan). Combining functional imaging in addition to anatomic imaging (especially in young patients and cases of paraganglioma) helps to identify multifocal lesions that may be missed with cross-sectional imaging alone and ensure that a nonfunctional anatomic abnormality is not removed instead of a “missed” functional lesion elsewhere. MRI and CT have sensitivity for pheochromocytoma of 98 and 89 %, respectively [11]. On CT, pheochromocytoma and paraganglioma can appear homogenous or heterogeneous and in addition to being solid, may be hemorrhagic, cystic, or necrotic compared to the usually homogenous and solid benign adenoma. Pheochromocytoma and paraganglioma have a higher density on CT than an adenoma and has a slower washout of contrast. Rarely, a pheochromocytoma can contain more lipid and be mistaken for an adenoma. On MRI, a pheochromocytoma is usually more signal intense, especially on T2-weighted imaging where approximately one third of the time it displays a “bright white light bulb” sign and has a slower washout phase [21]. An old adage is that a solid adrenal tumor that is bright on T2-weighted imaging is cancer or a pheochromocytoma until proven otherwise. Especially in younger patients or those with a higher suspicion for a genetic mutation, anatomic imaging from skull base to pelvis should be considered to look for paraganglioma .
The two main functional studies for pheochromocytoma and paraganglioma are the MIBG scan either with 123-iodine or 131-iodine and the PET scan [22]. MIBG is an analog of norepinephrine and is readily taken up by the norepinephrine transporter. The overall sensitivity for 131I-MIBG scanning is 77–90 % and the specificity is even higher at 95–100 %. Sensitivity improves with the use of 123I-MIBG and single photon emission computed tomography (SPECT) imaging to 88–96% [23]. However, the sensitivity of MIBG imaging is lower in cases of familial disease, malignant disease, abdominal paraganglioma, and nonfunctional head and neck paraganglioma. The sensitivity is especially low in cases of small, multiple paraganglioma, thus rendering MIBG less helpful with succinate dehydrogenase (SDH)-related disease. Further drawbacks of the MIBG study include the need for thyroid blockade, the potential need for withdrawal of catecholamine-blocking medications, limited image resolution, and long study time. In contrast, PET scanning has emerged as an extremely valuable tool for all cases of pheochromocytoma and paraganglioma, but especially for cases of multiple paraganglioma. Several different tracers have been utilized, most notably 18F-dopamine, 18F-DOPA, and 18F-flurodeoxyglucose (FDG). The sensitivity of 18F-FDG PET has been reported between 92 and 100 % and its specificity at 90 % [23]. The sensitivity of 18F-DOPA PET has been reported between 98 and 100 % with a specificity of 100 % [23]. 18FDG is the only tracer currently in wide use, and most pheochromocytomas and paragangliomas accumulate 18FDG (Fig. 3) .
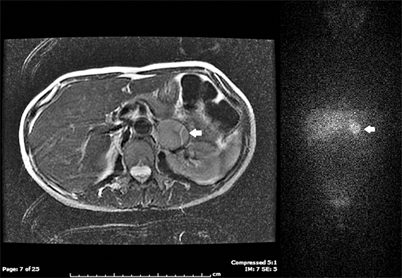
Fig. 3
T2 MRI and planar 131I-MIBG of a left pheochromocytoma ( white arrows). MRI magnetic resonance image, MIBG metaiodobenzylguanidine. (Images courtesy of Luisa Kropcho, M.D., and Ryan Restrepo, M.D., Department of General Surgery, Naval Medical Center Portsmouth, Virginia)
Adrenal tumors suspected of being a pheochromocytoma must not be biopsied as this may precipitate a “pheo crisis.” In fact, there are very few situations in which biopsy of an adrenal tumor is indicated. Perhaps the only scenario in which biopsy of an adrenal tumor is helpful is in the instance when a metastatic lesion is suspected and confirming the presence of an adrenal metastasis would change the management from surgical resection of the primary to systemic therapy.
Genetic Syndromes
Instead of the classic “10 %” rate of germ-line mutation, it is now recognized that at least 30–35% of pheochromocytomas and paragangliomas demonstrate a germ-line mutation [15, 24, 25]. In children presenting with pheochromocytoma or paraganglioma, this rate of germ-line mutation has been cited to be as high as 40% [7]. There are at least 17 causative genes, including RET, vHL, NF1, SDH ( A, B, C, D, and AF2), MAX, PHD2, K1F1Bβ, and TMEM127 [14, 15, 26] (Table 1).
Disease | Mutation | Predominant catecholamine | Usual location | Behavior | Associated findings |
|---|---|---|---|---|---|
MEN 2a | RET | Epinephrine | Adrenal | Benign | MTC, HPT, cutaneous lichen amyloidosis |
MEN 2b | RET | Epinephrine | Adrenal | Benign | MTC, mucosal neuromas, marfanoid habitus, Hirschprung’s disease |
Von Hippel-Lindau (type 2) | VHL | Norepinephrine | Adrenal (bilateral 40 %) | Benign | Retinal angiomas, CNS hemangioblastomas, pancreatic cysts and NET’s, renal cell carcinomas, epididymal cystadenomas |
Von Recklinhausen (Neurofibromatosis) | NF1 | Epinephrine | Adrenal | Benign (10 % malignant) | Neurofibromas, Café-au-lait spots, axillary and groin freckling, iris hamartomas, macrocephaly, mental retardation |
Familial paraganglioma syndrome (FPS) | SDHA | – | – | Benign (up to 14 % malignant) | Leigh disease, GIST, renal cell carcinoma, pituitary adenoma |
FPS type 4 | SDHB | Norepinephrine but can produce dopamine | Extra-adrenal abdomen, mediastinum, pelvis | Malignant | Renal cell carcinoma, papillary thyroid cancer |
FPS type 3 | SDHC | Can be parasympathetic. Can produce norepinephrine, dopamine | Skull base/ neck | Benign | Renal cell carcinoma |
FPS type 1 | SDHD | Can be parasympathetic. Can produce norepinephrine, dopamine. | Skull base/ neck | Multiple. malignant | Renal cell carcinoma |
FPS type 2 | SDH AF2 | – | Skull base/ neck | – | – |
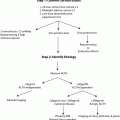
Genetic screening should be performed in patients with bilateral adrenal disease, paraganglioma, multifocal disease, and age younger than 50. However, given this higher rate of genetically linked disease, some groups advocate mandatory genetic testing for any patient with a pheochromocytoma or paraganglioma. Using the patient’s specific tumor characteristics combined with the information in Table 1, a directed, cost-effective testing strategy can be devised (Fig. 4).