Cox regression models
No age stratification
With age stratification
HR 95% CI
P
HR 95% CI
P
All patients (n = 60)
Recurrence (time dependent; yes vs. no)
2.7 (1.0–7.3)
0.05
4.4 (1.4–14)
0.01
Radicality at surgery (subtotal vs. total)
2.0 (0.9–4.5)
0.09
1.1 (0.5–2.5)
0.9
Radiotherapy (yes vs. no)
0.5 (0.2–1.2)
0.1
0.3 (0.1–0.8)
0.01
Patients surviving the first 6 months after surgery (n = 50)
Recurrence (time dependent: yes vs. no )
1.6 (0.5–4.7)
0.4
2.2 (0.6–7.6)
0.2
Radicality at surgery (subtotal vs. total)
1.6 (0.6–4.5)
0.4
0.9 (0.3–2.5)
0.8
Radiotherapy (yes vs. no)
1.4 (0.4–4.1)
0.6
0.7 (0.2–2.4)
0.6
Cardiovascular Mortality
Patients with CP have a 3–19 fold higher cardiovascular mortality in comparison to the general population [1, 9, 13]. The cohort from UK had the highest SMR of 19.4 (95% CI 8.08–46.7) for cerebrovascular deaths [9]. In the first Swedish cohort the cardiovascular (including cerebrovascular) mortality was three-fold enhanced (SMR 3.21, 95% CI 1.29–6.61) [1] and in a recent study the cerebrovascular mortality was 5.1 (1.7–12) [3]. Female gender seemed to be at particular risk for cardiovascular mortality (SMR 11.4 and 4.9, respectively) compared to male gender (SMR 4.79 and 3.2, respectively). This gender difference was also seen in the Dutch cohort with a higher SMR among females (SMR 3.84; 95% CI 1.47–7.22) compared to males (1.84; 95% CI 0.33–4.58) [13].
Morbidity in Craniopharyngioma
The long-term morbidity in patients with CP is substantial and is mainly based on the tumour location and size, recurrence rate, and its treatment. The morbidity includes hypopituitarism, hypothalamic involvement (obesity, thirst disorders, thermoregulatory disorders, somnolence and sleep apnoea, and cardiac arrhythmia), cardiovascular risk factors , visual and neurological problems, and reduced quality of life and cognitive function [6]. The treatment of a CP may be achieved using either primary gross total resection, which may increase the risk of hypothalamic, neurological, endocrine, and visual damage [6, 16], or partial resection of the tumour followed by CRT, as an effective means of preventing recurrences ( [17]).
Cardiovascular Morbidity
The increased cardiovascular mortality is also related to cardiovascular morbidity in CP patients. Childhood obesity due to CP is leading to severe atherosclerotic cardiovascular disease , type II diabetes mellitus, and metabolic syndrome [13, 18]. A study based on The Swedish National Patient Registry showed an increased risk for type II DM (six-fold) [3]. Recently, a total of 19 childhood onset CPs patients with hypothalamic tumour involvement agreed to participate in a CT scan investigation of the liver [19, 20]. A nonalcoholic fatty liver disease (NAFLD) was recorded in about 50% of CP patients with hypothalamic involvement which was associated with elevated liver enzymes and homeostasis model assessment index. NAFLD is considered a feature in the metabolic syndrome.
Holmer et al. [21] evaluated the prevalence of cardiovascular risk factors after long-term complete hormone replacement in childhood onset CP patients and showed increased cardiovascular risk factors in particularly CP women and in patients with hypothalamic involvement by the tumour [21]. Typical risk assessments for cardiovascular disease showed increased levels of hormone sensitive C-reactive protein and low density lipoproteins [21]. In addition, significantly more treatment for cardiovascular diseases (anti-hypertensive and anti-diabetes treatments and lipid lowering drugs,) and/or manifestations of the metabolic syndrome were present [21]. This is in accordance with another study also showing an increased prevalence of hypertension and of other cardiovascular morbidities in CP patients [13].
Hypopituitarism
Complete hypopituitarism is encountered in a majority of CP patients. At least three pituitary hormone deficiencies have been reported in 54–100% of patients with CP [22]. In a study from the Netherlands the long-term prevalence rate of total anterior pituitary insufficiency was 89% [13], and from Ireland the percentage of GH-, gonadotropin-, adrenocorticotropin-, and TSH-deficiency it was 91%, 93.5%, 92%, and 86%, respectively [11]. The prevalence of diabetes insipidus (DI) was 81% [11].
Pituitary deficiency per se and its treatment might through various metabolic effects contribute to the enhanced cardiovascular morbidity and mortality seen in epidemiological studies. However, panhypopituitarism is interrelated to recurrences of the CP, cranial radiotherapy, and with hypothalamic involvement by the tumour, thus it is almost impossible to identify hypopituitarism as an independent risk factor. GH deficiency is probably the most frequent pituitary hormone deficiency and associated with increased levels of cardiovascular risk factors [23]. GH therapy was shown to have beneficial effects on lean and body fat mass, total and low density lipoprotein cholesterol, and diastolic blood pressure in hypopituitary patients [24]. Only few studies have evaluated the effect GH therapy in CP patients and in comparison to patients with non-functioning pituitary adenoma, CP patients had higher prevalence of pituitary deficiencies, were more obese, and had more dyslipidaemia [25]. Two years of GH replacement showed the same effect on fat-free mass, lipids, but CPs were less likely to lose body fat, which is possibly a hypothalamic effect.
Also ACTH deficiency and glucocorticoid supplementation is of importance. This was highlighted by patients with Addison’s disease, who suffer from increased premature mortality in comparison to the general population [26]. In addition, high substitution doses of cortisone is associated with increased cardiovascular mortality in patients with previous acromegaly [27]. Subclinical hypothyroidism causes increased cardiovascular risk [28]. A study from Klose et al. [29] showed that low levels of serum free T4 were associated with higher BMI, waist, and hip circumference, but lower levels of the good -HDL- cholesterol.
Hypothalamic Obesity
The hypothalamus (HT) is a small, complex area of the brain and is involved in endocrine function and energy homeostasis. Hypothalamic damage due to tumour or its treatment is the most devastating consequence of a CP. Approximately 50% of children treated for CP are obese at follow-up and hypothalamic damage seems to be a major cause [30, 31]. The HT consists of distinct nuclei including the infundibular nucleus (INF) , the paraventricular nucleus (PVN) , the dorsomedial nucleus (DMN) , and the ventromedial nucleus (VMN) [32, 33]. The peripheral afferent hormones leptin, ghrelin, and insulin all have receptors located on neurons within VMH. Damage to these neurons leads to defective transduction of afferent signal of fat accumulation, satiety, and metabolism. In the postoperative phase of a CP destruction of these nuclei induces hyperphagia, hyperinsulinaemia, and weight gain [30, 33]. The resultant lack of anorexigenic pressure on the melanocortin receptor-4 results in increased feeding behaviour and energy efficiency (with reduced fat oxidation) to store energy substrate as fat. Overt hyperphagia is very prevalent in the first period after operation for about 12 months [34] but seem to be conspicuously absent in the long-term follow-up of these patients [35, 36].
Adipose tissue is richly innervated by sympathetic nerve fibres that control lipolysis. Lipogenesis is also controlled by parasympathetic innervation of adipose tissue originating from separate sympathetic and parasympathetic neurons in the periventricular nucleus (PVN) and supra chiasmatic nucleus (SCN) [37]. Thus, SCN has a unique position to balance both branches of the autonomous nervous system. A large proportion of the CP patients have supra sellar involvements and thus damage to the SCN with an altered regulation of central clock mechanism, which predispose to alterations in metabolism [14, 37]. A disturbance in the circadian rhythms may cause daytime sleepiness demonstrated in childhood onset CPs patients with hypothalamic obesity, which correlate with low morning and nocturnal melatonin levels in saliva [38]
The primary pathogenesis of hypothalamic damage seems to start with hyperinsulinaemia, due to destruction of the VMH nuclei causing imbalance of autonomic nervous system, resulting in suppression of the sympathetic nervous system and stimulation of the vagus [33]. Indeed CP patients have hyperinsulinaemia, with increased levels in the relation to tumour growth (Fig. 1, [36]). Hyperinsulinaemia increases lipogenesis in liver and adipose tissue and also lipoprotein lipase activity accelerating endogenous (very low density lipoproteins and triglycerides) lipid production [32]. Fat mass is accumulated with an increase in weight and BMI [32].
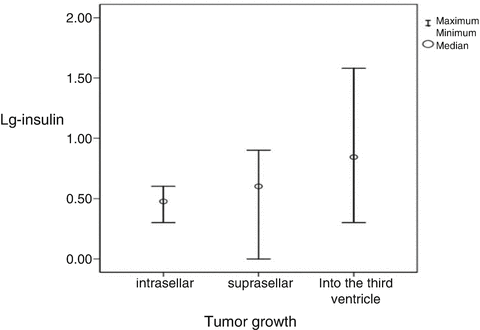
Fig. 1
Correlation between tumour growth and the logarithm for serum insulin for all patients, r = 0.57, P < 0.001; for men, r = 0.53, P = 0.011; and for women, r = 0.66, P = 0.002 [36]
There is a high degree of correlation between total adiposity and leptin levels in obese CP patients [33] similar to levels found in normal obese subjects, but among the CP patients higher leptin/kg fat mass were found, suggesting lack of sensitivity to endogenous leptin. Elevated leptin levels in relation to BMI, thus leptin resistance , have been recorded in both children [39] and adult CP [36] with hypothalamic involvement.
The gastric hormone, ghrelin , is a peripheral produced and centrally active orexigenic hormone that is considered to be an important gut-brain signal for appetite control and energy balance. It has been proposed that ghrelin acts as an initiator for food consumption in humans, with a pre-prandial rise and a post-prandial fall in plasma [40]. In obese normal individuals there is an inverse relation between ghrelin and obesity and BMI [41], which is also confirmed in CP patients [36]. Further, in CPs there is an inverse correlation between plasma ghrelin with tumour growth into the hypothalamus [36]. Previously a weaker post-meal ghrelin response was shown in obese CP patients with hypothalamic involvement, probably due to insulin resistance [42]. It is possible that a low ghrelin response together with uncoupling of feeding-related signals such as leptin with sympathetic activity on the weight regulatory loop reinforces obesity in acquired HO and not hyperphagia. In addition, chronic pro-obesity effects of vagally induced hyperinsulinaemia contribute to persistence of obesity in adulthood [43]. A cross talk between signaling events downstream of insulin receptors and leptin receptors occurs and thus cross-desensitisation between insulin and leptin pathways can occur. Insulin resistance at the intracellular signaling levels causes a relative resistance to leptin thereby enabling the body to tolerate new adiposity. And indeed CP patients with HO have relative hyperinsulinaemia for their obesity and with a significant correlation to tumour growth [36].
Importantly a reduction in energy expenditure has been recorded in a study of children with hypothalamic obesity (HO) suffering from tumour in the hypothalamic region, i.e. CPs [44]. Pediatric and adult patients with childhood onset CPs were found to have a lower resting energy expenditure when compared to controls [45]. Further, Holmer et al. [36] have shown that adults with childhood onset CP had significantly lower basal metabolic rate (BMR) compared to controls. After adjustment, patients had lower BMR compared to controls (−90 kcal/24 h, P = 0.02), and the ratio (Energy Intake) EI/BMR was significantly lower in patients with hypothalamic damage (Fig. 2). The total caloric intake was not significantly different between CP patients and controls. This accords with a self-assessment by nutritional diary from a previous study on childhood CP and BMI matched controls [35]. Further, Holmer et al. [36] also showed that similar dietary macronutrient composition was found in patients and controls, and interestingly a lower EI (1778 vs 2094 kcal/24 h; P =0.008) was recorded among patients with hypothalamic involvement by the tumour. With the use of questionnaire only significantly higher scales in restricting food intake, but no difference in disinhibition or hunger, were recorded in patients [36]. In addition, reduced physical activity compounded by neurological deficits and visual disturbances worsens the obesity in these patients [35]. Harz et al. [35] suggested that reduced physical activity rather than increased energy intake is responsible for the obesity in these subjects.
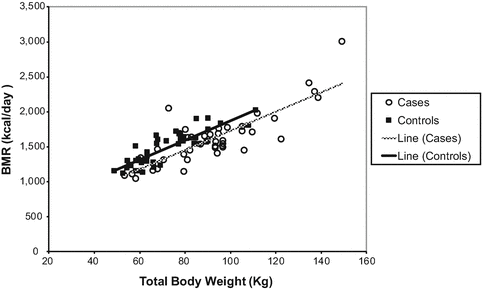
Fig. 2
Linear association between total body weight (kg) and BMR (kilocal/day) among patients and controls. Patients had significantly lower BMR compared with controls after adjustment for sex and total body weight in linear regression analysis (mean difference, −90 kcal/24 h; 95% CI, −160 to −10; P = 0.02) [36]
Recent data show that hypothalamic involvement was associated with lower overall survival at diagnosis and follow-up when compared with sellar mass without hypothalamic involvement [46]. Hoffmann et al. [19] analysed eating disorders and behaviours of 101 childhood onset CPs survivors and 85 BMI matched controls. Severely obese patients presented with pathological eating behaviours and more weight problems than obese and normal or overweight patients. However, childhood onset CPs with different degrees of obesity showed similar or even less pathological findings. The conclusion was that although disturbed eating was present this was not the root cause of their hypothalamic obesity [19, 20]. Roth et al. assessed the pre- and post-meal response to visual food cues in childhood onset CPs’ brain regions of interest using functional magnetic imaging (fMRI) [47]. Following a test meal, BMI-matched controls showed suppression of activation by high-caloric food cues while in childhood onset CPs patients showed trends toward higher activation. These altered perceptions of food cues indicated on fMRI, especially after food intake, support the body of evidence that disturbed eating behaviour is not the root cause of obesity in childhood onset CPs with hypothalamic obesity.
Bone Health
The hypothalamus regulates bone and adipose tissue via a complex and fine-tuned interplay of endocrine mechanisms (of which neuropeptide Y is a key regulator) together with the sympathetic nervous system [48]. Obesity in healthy subject is protective against low BMD [49] possibly through increased leptin levels [50]. However, in obese CP patients with hypothalamic damage leptin levels were shown to be higher in relation to body mass index, indicating leptin resistance [39], and among adolescents leptin resistance was shown to be negatively correlated to BMD [51]. In animal models leptin resistance has been correlated to bone loss [52].
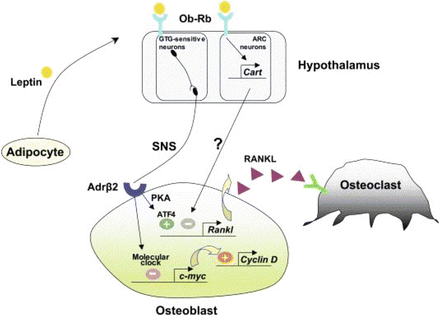
Leptin binds to its hypothalamic receptors and induces two cascades to control bone mass ([52], Fig. 3). In the arcuate nuclei, it increases Cart expression that in turn regulates, via an unknown mechanism, Rankl expression by osteoblasts, and thereby bone resorption. Leptin also binds to receptors on the GTG-sensitive neurons of the VMH nuclei, inducing an increase in sympathetic activity which signals to osteoblasts via the β2 adrenergic receptors present at their surface. Two distinct molecular cascades downstream of this receptor are then activated. One inhibits osteoblast proliferation via the molecular clock regulation of c-myc and Cyclin-D expression. The other, mediated by PKA phosphorylation of ATF4, promotes Rankl expression and thereby bone resorption [52]. Upon inactivation of the molecular clock, the leptin/SNS arm regulating osteoblast proliferation via c-myc and Cyclin-D is overactivated, leading to an increase in bone formation and thus a high bone mass phenotype. Cart absence leads to a decrease in bone mass because of an isolated increase in osteoclast activity following Rankl overexpression by osteoblasts [52].