Fig. 1
Histological specimens of adamantinomatous craniopharyngioma demonstrating epithelial regions admixed with microcysts and a reactive glial surround: (a) Haematoxylin and eosin stain; (b) Immunohistochemistry for β-catenin demonstrates nuclear and cytoplasmic staining in the epithelial cells without uptake in the glial or cystic regions of the tissue
Increased expression and/or activation of β-catenin likely leads to active expression of multiple target genes. These genes include Lef1, Axin1, c-myc, and CyclinD1, which is a major regulator of cell-cycle progression to the proliferative stages [5, 10, 11, 16]. Axin2, a well-recognised inhibitor, and BMP4, which plays a critical role in tooth development and increases cell proliferation in conjunction with β-catenin accumulation, are also target genes with increased expression as a result of increased Wnt signaling [9, 12, 16]. Both Axin2 and BMP4 were shown to co-localise to the nucleus with β-catenin with increased mRNA levels [9, 12, 13, 15]. Lef1, an enamel protein, expression is activated by BMP4 [15, 16].
ACP Genome Analyses
CTNNB1 Mutation
Exon 3 of the CTNNB1 gene (β-catenin) encodes a degradation targeting motif. Mutation may confer resistance of the protein to the usual destruction complex [11, 16] and manifest through nuclear accumulation of β-catenin [5, 7, 10, 11, 19]. The first descriptions of this genome level aberration in ACP demonstrated CTNNB1 mutations in 69–100% [6, 16, 20] of ACP specimens and the absence of such mutations in PCP and other parasellar tumors [6, 16]. Using massively parallel sequencing of 12 specimens of ACP, followed by targeted genotyping in an additional 53 specimens CTNNB1 mutation, were identified in 95% (11/12 and 51/53) of specimens [21]. CTNNB1 mutations were absent in PCP. Other groups have identified CTNNB1 mutations in approximately 70–80% of ACPs [12, 16, 19, 22]. Although the source of thus variation remains unknown, potential explanations could include intratumoral heterogeneity, sampling error, or even the existence of multiple subtypes of ACP.
As described above, the majority of CTNNB1 mutations in ACP are upregulating and involve phosphorylation of serine or threonine sites encoded by exon 3, which ultimately encode the degradation targeting box of β-catenin, which corresponds to the GSK3β binding domain [5, 7, 9, 11–13, 19, 20]. Mutations of CTNNB1 have been identified at a number of different codons [9, 15, 16]. These include codon 32, which is adjacent to the phosphorylation site of GSK3β, as well as codons 33, 37, and 41, which affect the serine and threonine residues that are targeted by GSK3β [6, 9, 14, 15, 19].
The presence of the CTNNB1 mutation in ACP may offer insight into the etiology of this tumor. Mutations of the GSK3β binding domain of CTNNB1 have been identified in other lesions, including calcifying odontogenic cysts and pilomatricoma [7, 16, 19], a benign skin tumor that is histologically characterised by anucleate squamous cells, similar to ACP. As such, ACP shares genetic and histological characteristics with tumors of odontogenic origin. However, it has been postulated that ACP arises from undifferentiated anterior pituitary epithelial stem cells and differentiates toward an oral epithelial phenotype [18, 20, 23, 24]. One model proposes that there is a small population of mutated cells, which express nucleocytoplasmic β-catenin, which are histologically identified within well-demarcated clusters [17]. This theory is supported by recent in vitro and animal models that are discussed in Chap. 2 and 3. Briefly, a leading hypothesis regarding ACP tumorigenesis is that para−/autocrine signaling emanating from this small population of CTNNB1 mutated cells (i.e. cancer stem cells) and that this drives tumor growth and invasion. It has also been postulated that microRNA derangements also in part help drive tumor behaviour [7]. It may therefore be that, while the tissue of origin differs, the phenotypic maturation of ACP tissue converges with that of odontogenic lesions [25, 26].
Epidermal Growth Factor Receptor
Although genome level dysregulation of the epidermal growth factor pathway does not appear to exist in ACP, there is evidence that upregulation of this pathway, potentially by epigenetic mechanisms, contributes to ACP cell growth and infiltration.
The epidermal growth factor receptor (EGFR) is a 170 kDa receptor tyrosine kinase (RTK) that is also known as HER1, ErbB1, mENA, and PIG61 [10, 18]. It is a transmembrane glycoprotein composed of an extracellular domain that is involved in ligand binding, a transmembrane portion, and an intracellular portion that makes up the tyrosine kinase domain and is the site for autophosphorylation [18]. EGFR can be activated by multiple growth factors, including epidermal growth factor (EGF), TGF-α, amphiregulin, epiregulin, heparin-binding EGF (HB-EGF), and betacellulin [18]. When the ligands bind the extracellular domain, the receptor undergoes dimerisation, which activates the intracellular tyrosine kinase domain. This results in phosphorylation of multiple tyrosine residues (e.g. Y992, Y1045, Y1068, Y1086, Y1148, and Y1173) [11, 18]. Binding of the ligand to the extracellular domain also promotes recruitment of other enzymes and proteins, which act as signal transducers and can lead to initiation of multiple intracellular cascades involved in regulation of cell proliferation, differentiation, apoptosis, and motility [18]. Alterations in EGFR signaling have been noted in multiple human tumor types [20] resulting from various mechanisms including EGFR gene amplification, activating mutations, or overexpression of ligands and receptors [18]. In some tumor types, a common location for mutation is the area that encodes the tyrosine kinase domain of EGFR, between exons 18 and 21 [18].
The mechanism of EGFR pathway contribution to ACP behaviour may lie in the interaction between EGFR and β-catenin [12, 18, 27]. When activated and phosphorylated, one interesting proposed mechanism is that EGFR co-localises with nuclear β-catenin and fascin, particularly at the infiltration border of ACP, within whorl-like cell clusters [12, 18]. As described above, in ACP, these clusters have been associated with activation of the canonical Wnt signaling pathway. This suggests a convergent role for both the EFGR and Wnt pathways in the pathogenesis of ACP [18]. This concept is supported by laboratory data [10, 20] and culture models that correlate EGFR pathway activation with ACP growth and migration of ACP cells [18]. Additionally, EGFR appears to be involved in the regulation of stem cell like properties in ACP through the regulation of the expression of stem cell markers and the activation of the EGFR pathway in β-catenin accumulating cells [5]. Lastly, the ACP transcriptome demonstrates high levels of EGF pathway genes, including AREG, EGFR, and ERBB3 [20].
SHH
Sonic hedgehog (SHH) plays an integral role in the maintenance of stem cell milieus in adult tissues [28] and in the normal development of several organs, including Rathke’s pouch [12]. The SHH pathway is involved in pituitary formation during early embryogenesis. The transmembrane protein Patched 1 (PTCH1) receptor complex binds the hedgehog ligand, which activates the pathway [22]. Once the ligand binds to the receptor complex, the frizzled class receptor, smoothened (SMO), is freed from PTCH1 inhibition. It then activates glioma-associated oncogene family zinc fingers—the GLI1, GLI2, and GLI3 transcription factors [22]. Pathological upregulation of SHH signaling contributes to mitogenic behaviour in numerous neoplasms, including glioma, basal cell carcinoma, lung cancer, and breast cancer [28, 29]. Among pediatric brain tumors, the most widely known example of hedgehog dysregulation is in the SHH subgroup of medulloblastoma [1].
As described in Chap. 2, expression of SHH has been observed in a murine model of ACP and in human ACP. In both mouse and human tissues, expression of SHH has been shown to co-localise in cells that have distinct nuclear localisation of β-catenin [28]. The pattern of expression, combined with our understanding of the SHH pathway in normal human pituitary development, supports the hypothesis that both autocrine and paracrine SHH signaling may contribute to ACP tumorigenesis. The importance of SHH signaling in the pathogenesis of ACP has been supported by two studies of the human ACP transcriptome, both of which have demonstrated significant upregulation of SHH pathway genes [20, 22] [Fig. 2]. In addition, a very recent study comparing the transcriptomes of ACP with PCP showed significantly higher expression of SHH pathway genes Axin2, Gli2, and PTCH1 in ACP [25]. This important finding further supports the possibility of WNT and SHH signaling pathways converging to support the growth of ACP. This study also showed that methylation profiling patterns are distinct between ACP and PCP, further establishing that ACP and PCP are indeed biologically distinct lesions with different mechanisms of pathogenesis.
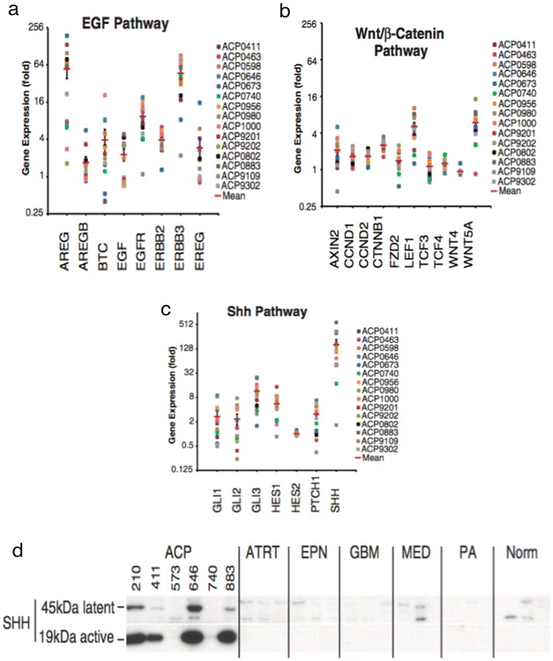
Fig. 2
Expression of the indicated developmental and cancer-related genes in individual ACP samples: (a) Epidermal growth factor genes; (b) WNT pathway; (c) Sonic hedgehog pathway; (d) Western blot analysis demonstrating overexpression of the latent pre-forms and active cleaved protein isoforms of SHH in ACP relative to other common pediatric brain tumors and normal brain. AT/RT atypical teratoid/rhabdoid tumor, EPN ependymoma, GBM glioblastoma, MED medulloblastoma, PA pilocytic astrocytoma, Norm, normal brain). Adapted from Gump et al. [20]
Additional Potential Relevant Pathways
In addition to the activation of EGFR and SHH pathways that we describe above, other molecules and pathways have been demonstrated to be uniquely expressed in craniopharyngioma, but are not yet as well described. As effective therapy for any brain tumor, including ACP, is likely to require multiple medications, further exploration of these pathways has clinical relevance.
Two studies of the ACP transcriptome found upregulation of certain matrix metallopeptidases [20, 30]. Gong and colleagues identified elevation of MMPs 2, 3, 7, and 9 in recurrent ACP, relative to primary ACP. Gump and colleagues [20] identified elevated levels of MMPs 9 and 12 mRNA transcripts in pediatric ACP, relative to other brain tumors and normal brain. The clinical relevance of these findings is increased by the availability of the oral MMP9/12 inhibitor, AZD1236. The latter group additionally identified the upregulation of three targets of the oral tyrosine kinase inhibitor dasatinib (EPHA2, LCK, and SRC), thereby revealing another potential therapeutic option [20].
Elevated levels of p63 have been identified in both ACP and PCP. P63 , also known as p51, p40, p73L, or KET, is a gene homologue of the p53 tumor suppressor family. The gene has two separate promoters, which result in different isoforms, one with an N-terminal transactivation, and one that lacks this N-terminal domain. These two isoforms have opposite functions and are responsible for cell-cycle arrest and proliferation [19]. Using immunohistochemistry, p63 nuclear immunopositivity was identified in both ACP and PCP [10, 19]. Evidence of p63 expression was also identified in the ACP transcriptome [20]. While p63 is suggested to play a key role in the control of epidermal proliferation and differentiation, impaired p63 expression has also been recently seen in squamous cell carcinomas arising in different organs and has been suggested to influence neoplastic cell transformation [19].
Papillary Craniopharyngioma
Distinction from ACP
As described above, the distinct mutations identified in ACP and PCP, the different transcriptome profiles, and the differences in genome wide methylation patterns support the view that ACP and PCP are biologically distinct entities with different mechanisms of pathogenesis. These genomic studies support the earlier findings from histological studies that demonstrated nuclear and cytoplasmic accumulation of β-catenin in ACP whereas β-catenin localised exclusively to the cell membranes of tumor cells in PCP, similar to the pattern of localisation in other CTNNB1 wild type tumors of the sellar region [10, 12, 16, 19].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


