Gene amplification
Insertions
Loss of heterozygosity
Minor alleles
Moveable elements
Mutations
Single-nucleotide polymorphisms
Deletions
Splice site variants
Tri-nucleotide repeats
3-dimensional chromatin domain
Chromatin folding
DNA methylation
Histone modification
Noncoding RNA
Nuclear matrix attachment/loop regulation
Transcription factor binding
Chromosome level
Aneuploidy
Polyploidy
Chromosome fragmentation
Defective mitotic figures
Micronuclei
Multiple nuclei
3D-telomere behaviour
Double minute chromosomes
Inversions
Large region amplification
Deletions
Translocations
Copy number variation
Cryptic translocations
Heterogeneity at the suborganismal level is underappreciated as most individuals are actually somatic mosaics (Iourov et al., 2008; Heng et al., 2011a, b). Known heterogeneity at the cellular level occurs in B cells and T cells following VDJ recombination. Likewise, different cell and tissue types each have unique epigenetic marks. Less appreciated is the effect of heterogeneity, including genome level heterogeneity in multiple normal tissues including brain and liver, and in the transition from normal physiological to pathological conditions. The key here is that throughout the life of an organism GEH increases, in particular during the formation of diseases. Thus it is important to distinguish germline and somatic cell heterogeneity, as well as normal development and disease conditions. Germline heterogeneity is heritable within the population whereas somatic heterogeneity is heritable only within the organism. In contrast to somatic cells, genome level heterogeneity is purified by sexual reproduction in each generation (Heng, 2007a, b; Wilkins and Holliday, 2009; Gorelick and Heng, 2011).
GEH in Cancer
The current view in cancer research is that heterogeneity can be eliminated using large sample sizes, despite the realisation that heterogeneity is an unavoidable key feature of cancer (Heppner, 1984; Heng et al., 2006a, b, c; Davidoff, 2009). This view has been cemented by molecular genetics and arises from successes in identifying disease gene loci and the hope that specific gene mutations can be targeted in cancer. Cancer progression is thought to occur in a stepwise clonal fashion (Hanahan and Weinberg, 2000; Vogelstein and Kinzler, 2004). New results demonstrate just the opposite; cancer is far from a stepwise, clonal process (Heng et al., 2006a, b, c). Furthermore, confusion exists as to what exactly constitutes genomic instability. Genomic instability is often regarded as any change from normal that is clonal, however instability implies that the genome is constantly in flux and therefore exhibit little clonality (at the genome level) (Heng et al., 2006a, b, c).
GEH occurs in cancer at all three levels of genetic organisation, that is, the genetic, epigenetic and most importantly the genome level (Table 1). Genome level change is ubiquitous in solid tumours though until recently there has been little appreciation for genome level heterogeneity, despite the fact that genome change occurs at a frequency on an order of magnitude higher than gene mutations (Bayani et al., 2007; Lupski, 2007; Heng et al., 2010a, b, 2011a, b). Instead focus has been placed on the identification of discrete molecular changes, such as the accumulation of APC, RAS and TP53 mutations in colorectal cancer. Interestingly it is more common for a colorectal tumour to not contain mutations in any of these three ‘driver’ genes than for a tumour to have mutations in all three (Wood et al., 2007).
Genome level heterogeneity
Genome level heterogeneity exists in multiple forms at the chromosomal level including, chromosome translocations, aneuploidy, as well as less well-known forms such as chromosome fragmentation defective mitotic figures, micronuclei and multiple nuclei and at the subchromosome level including large-scale copy number variation, inversions and three-dimensional (3D) telomeric behaviour (Table 1) (Heng et al., 2004; Shen et al., 2005; Duesberg, 2007; Stevens et al., 2007, 2010, 2011; Ye et al., 2007; Mai, 2010). Genome level change has long been associated with cancer, however research has focused on detection of clonal events (Nowell, 1976), as cancer cytogenetic studies are typically only interested in clonal change (Mitelman, 2000). Nonclonal chromosome aberrations (NCCAs) are pervasive in tumours and drive cancer progression (Heng et al., 2010a, b, 2011a, b). Longitudinal studies of chromosome change in models of cancer progression have shown that genome level heterogeneity waxes and wanes. Genome level heterogeneity increases over time, promoting the evolutionary potential, until the population encounters a selection event, which eliminates a large portion of the heterogeneous population that cannot survive such conditions. The remaining population then expands clonally while slowly acquiring more GEH. This cycle occurs repeatedly throughout the lifespan of a tumour. Following selection low levels of genome instability remain present. If the system is subjected to further pressure, genome heterogeneity is rapidly regained. This has been confirmed in recent reports of extensive genome chaos where large-scale genome change takes place during cancer progression (Heng et al., 2010a, b, 2011a, b; Stephens et al., 2011; Stevens et al., 2011). Without the effects of genomic heterogeneity, the tumour system would lack robustness to outcompete normal cells.
Genome level heterogeneity is the primary level of instability that drives cancer progression (Figure 1) (Heng et al., 2006a, b, c). The large-scale expression change that takes place due to genome change directly affects the function of protein networks. Identification of a cyclical pattern of genome stability and instability has led to the establishment of the evolutionary mechanism of cancer, which is equal to the sum of all potential molecular mechanisms of cancer (Heng et al., 2006a, b, c, 2010a, b). This concept led directly to the synthesis of the genome theory of cancer evolution (Heng, 2009), which states that stochastic genome level changes drive cancer progression in most cases. In limited cancer types, primarily in haematological malignancies, activation of strong oncogenic pathways can drive cancer progression, and in these cases heterogeneity is more limited throughout the tumour, in particular during earlier more treatable stages. Even in these cases there can be significant genome heterogeneity, especially as drug resistance is acquired.
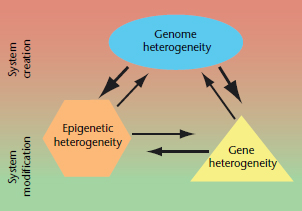
Figure 1 Interactions between the three levels of genetic and epigenetic heterogeneity (GEH). Each of the three levels (genome, gene and epigenetic) of GEH can impact the others, however genome heterogeneity has the largest effect of the three. GEH drives somatic evolution and each of the three layers contributes to evolution. Genome heterogeneity is the primary contributor to macroevolution which creates new systems, whereas epigenetic and gene heterogeneity primarily contribute to microevolution which modifies existing systems.
Discovery of dominant oncogenic pathways early in the molecular era of cancer research has driven the field to search for common stepwise patterns that drive cancer progression. For example, identification of the Philadelphia chromosome and the resultant BCR–ABL fusion in chronic myelogenous leukaemia (CML) has set the stage for attempts to identify common targetable events in other types of cancer (Rowley, 1973). Despite successful identification of an oncogenic fusion protein associated with CML, questions still exist as to the true role of the fusion protein in carcinogenesis. For example, some normal individuals also have detectable levels of the fusion protein in lymphocytes. The bcr–abl fusion protein that is thought to drive CML can be targeted with the drug imatinib. Failure of imatinib therapy has also been linked to other genome level changes other than the specific mutation of the BCR–ABL gene (Druker et al., 2006). Furthermore, after blast crisis imatinib is of little utility when there is genome chaos.
Gene level heterogeneity
It is generally accepted that the sequential accumulation of mutations leads to cancer (Vogelstein and Kinzler, 2004). This is the basis for sequencing the cancer genome as promoted by multiple consortiums. However, scores of sequencing results demonstrate that gene mutations are highly stochastic. Single cell sequencing has demonstrated that loci are randomly mutated in cancers (Bielas and Loeb, 2005). Others have shown different clonal expansions in different areas of tumours. Whole-genome sequencing (WGS) of tumours has demonstrated extensive heterogeneity in the same types of tumours from different patients (Wood et al., 2007; Hayden, 2010; Berger et al., 2011). The overwhelming message is that there are no commonly shared mutations amongst individual tumours, but instead mutational heterogeneity is quite common within tumours and between patients with the same type of tumour.
Epigenetic heterogeneity
Studies of epigenetic change focus on four principle levels in cancer including change in deoxyribonucleic acid (DNA) methylation, histone modification, noncoding ribonucleic acid (NC-RNA) and in 3D DNA interactions (Table 1). DNA hypomethylation commonly occurs in cancer, and was the basis of the identification of epigenetic regulation (American Association for Cancer Research Human Epigenome Task Force and The European Union, Network of Excellence, Scientific Advisory Board, 2008). The epigenetic field has risen rapidly in response to the failure of finding shared gene mutations that drive cancer. The reasoning for this is that if gene level change cannot explain cancer progression, the regulation of those genes can. This logic is problematic however, as epigenetic regulation is amazingly complex and dynamic (Sottoriva et al., 2011). The effects of epigenetic complexity is likely not explained by its association with specific genes, and the surface of the complexity of the epigenome has only been scratched.
DNA methylation patterns can be heterogeneous between different tissues, as it is between individuals. Across individual tumours DNA methylation can be extensively heterogeneous. For example, in hepatocellular carcinoma, comparison of methylation patterns between tumours, adjacent liver cells and healthy liver cells shows that tumours are less related than normal cells (Tao et al., 2011). The meaning of this heterogeneity however is contested as is DNA methylation in general. For instance, despite heavy methylation of the gene, Hypermethylated in Cancer 1 (HIC1), it can be strongly expressed. DNA methylation is heritable, though its patterns can change (Jenal et al., 2009). In contrast, histone modification is not (American Association for Cancer Research Human Epigenome Task Force and The European Union, Network of Excellence, Scientific Advisory Board, 2008).
Histone modifications are a second facet of epigenetic regulation (American Association for Cancer Research Human Epigenome Task Force and The European Union, Network of Excellence, Scientific Advisory Board, 2008
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


