FIGURE 1 | Cell type specificity of DNA methylation. This figure shows the dissimilarity of cell types based on their methylation frequencies from targeted bisulfite data that covered CpG islands on chr12 and chr20. Cell types that are closer together share a more similar methylation signature. The fibroblasts, PGP1F (Personal Genome Foundation 1 Fibroblast), BJ, IMR90, and hFib2 (human Fibroblast), cluster closely together, while the lymphoblasts PGP9L (Personal Genome Project 9 Lymphoblast), PGP3L, and PGP1L also cluster together closely. Regarding the pluripotent cell lines, the ES cells (Hues12, Hues42, and Hues63) cluster very closely together. The hybrid cell line, which consists of fused nuclei from Hues6 and BJ, also clusters closely with the ES cell group. The induced pluripotent stem (iPS) lines appear to be much more similar to the ES cell group than the differentiated fibroblasts and lymphoblasts but the iPS group exhibits a wider spectrum of methylation signatures than the ES cell group or the two differentiate cell groups.
DNA Methylation as a Clinical Biomarker
DNA methylation can classify cell types into subpopulations, which can exhibit unique phenotypes. One cancer study examined the methylation profiles of blast cells taken from 344 diagnosed acute myeloid leukemia (AML) patients.42 Clustering these methylation profiles created 16 unique AML subtype clusters. Three of those patient clusters were defined by the WHO classification,43 eight were enriched for specific genetic or epigenetic lesions, and the remaining five could not be explained by current knowledge; all of these subtypes were distinct when compared to normal bone marrow cells. The authors used the methylation signatures of 18 methylation probe sets that covered 15 genes and developed a classifier that predicted the overall survival and event-free survival of an AML patient (P-value < 0.001, multivariate Cox proportional hazards model). These authors showed that DNA methylation signatures can act as biomarkers that foretell patients’ clinical outcomes. Another study focused on breast cancer and used methylation signatures to distinguish between the different breast cancer mutation types (BRCA1, BRCA2, BRCAx).44 The authors report that DNA methylation profiling predicted BRCA1, BRCA2, and BRCAx tumors with error rates of 11, 31, and 36%, respectively. Classification based on DNA methylation signatures was significantly more accurate than using gene expression data, which resulted in error rates of 11, 44, and 71%, respectively. However, the gene expression data were able to cluster the breast cancer samples into intrinsic subtypes (i.e., basal, luminal A, luminal B, HER2-amplified, and normal-like) while the DNA methylation data could not.45 Another group used the DNA methylation signatures from over 300 peripheral blood samples as an indicator for ovarian cancer.46 Methylation studies involving colorectal cancer and breast cancer have shown that peripheral blood samples can indicate the presence of cancer when compared to healthy controls.47–51 Starting with 25,642 CpG sites, the authors found that a 100 CpG sites in peripheral blood cells that accurately discriminated between healthy and pretreatment ovarian cancer patients. Using these selected 100 CpG sites, the authors were able to correctly identify ovarian cancer samples (58 healthy samples and 43 pretreatment cases) with an area under the receiver operating characteristic (ROC) curve (AUC) of 0.82. AUC describes the ability of a test to detect true positives versus false positives. Tests with AUC values close to 1 represent high true positive and low false positive detection rates, and AUC values close to 0.5 represent discriminatory power that is similar to random selection. Tests with AUC less than 0.5 perform worse than random selection. Comparing a sample post-treatment population that showed signs of active disease with one that served as a healthy control, the classifier correctly identified those active disease patients with an AUC of 0.76. Finally the classifier was able to discriminate between post-treatment samples showing active disease and post-treatment samples without active disease (0.74 AUC). Additional tests showed the CpGs used in the classifier were not dependent on age. DNA methylation can be used to predict cancer-related phenotypes as seen in these three studies.
INTRACELLULAR DNA METHYLATION: ALLELE SPECIFIC INSTANCES
A mammalian cell contains a paternal and a maternal chromosome set. Most biological activity is thought to be symmetrical across the paternal and maternal chromosomes, but there are regions where each parental allele is regulated uniquely. Allele-specific methylation, ASM, (Figure 2a) is a common feature of these regions and ASM can be coupled with allele-specific expression, ASE (Figure 2b). Imprinted genes consist of genes exhibiting ASM and ASE in a parent-of-original specific manner. ASM near the loci of these ASE genes reveals significant methylation differences, where one allele is significantly more methylated than the other. There are between 50 and 100 known imprinted genes in mouse and human.52–54 However, over 1000 genes have been found to exhibit parent-of-origin allelic gene expression effects in the mouse brain.55 Besides imprinting, ASM can also occur in a manner that is independent of parental origin. This occurs in chromosome X inactivation randomly, but it has also been unexpectedly observed in numerous autosomal regions.39,56 This section explores ASM and places it in the context of other regulatory mechanisms that co-occur at these sites.
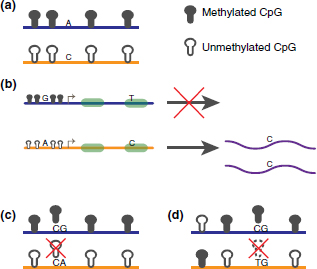
FIGURE 2 | ASM examples. (a) A schematic of typical ASM. The two alleles are represented separately as blue and orange lines. A SNP distinguishes the alleles and the blue allele is methylated while the orange allele is not. This is considered an ASM region. (b) This figure shows a gene whose promoter region is methylated in the blue allele and unmethylated in the orange allele. A SNP site at the promoter distinguishes the alleles. Additionally, another SNP site in an exon differentiates the alleles. This figure shows how ASM can be used to predict ASE behavior. Additional haplotyping can associate the SNPs thereby allele specifically linking the methylation status of a regulatory region to its expression. (c) An instance of a SNP overlapping with a CpG site in an ASM region. The SNP in this example disrupts a CpG site such that the CpG site no longer exists in the orange allele. The ASM behavior seen in this region may be caused by the SNP itself or the SNP/CpG overlap. (d) An instance of a SNP overlapping with a CpG site in a non-ASM region. Similar to (c), the overlapped CpG site no longer exists in the orange allele.
Chromosome X Inactivation
The extensive research into chromosome X inactivation (XCI) has illuminated a pathway for allele specific methylation via noncoding RNAs (ncRNAs). To maintain proper dosage specificity in female cells, one chromosome X copy is inactivated upon differentiation. The inactivated chromosome is heavily methylated, compacted into a heterochromatic state, and is largely transcriptionally silent. Experiments have shown that this process is directed by large noncoding RNAs, mainly Xist and TsiX, which are regulated at the X inactivation center.52,57–62 Prior to XCI, the two X chromosomes pair at and around the X inactivation center.63–65 The pairing and XCI initiation rely on the binding of Oct4 and the CCCTC-binding factor (CTCF) at the XCI region.63,66,67
Changes in the allele-specific transcription of TsiX and Xist loci initiate a cascade that results in the methylation-mediated silencing of one copy of chromosome X via a yet poorly understood counting mechanism.63,68,69 Transcription of Xist is up regulated, while TsiX is down regulated on the selected Xi chromosome (inactive chromosome X); whereas on Xa (active chromosome X), Xist is down regulated, and TsiX is up regulated. On Xa, TsiX has been shown to associate with Dnmt3a at the Xist promoter region. This leads to the de novo methylation of the Xist promoter and thus the silencing of Xist transcription on Xa.70 Xist contains a RepA element to which PRC2 binds and this interaction recruits PRC2 to chromosome X.71,72 As Xist transcription remains high on Xi, Xist localizes to various locations across Xi and its recruitment of PRC2 leads to a chromosome-wide H3K27me3 modification of nucleosomes on Xi.62,73 PRC2 then likely recruits DNA methyltransferases to Xi, where it is globally methylated.74 The XCI process reveals an RNA directed DNA methylation (RdDM) pathway in human. Experiments have observed active RdDM pathways in Arabidopsis, but there are few such mammalian examples.2,75–78
Imprinted Genes
Imprint control elements (ICE) regulate the allele specific expression of imprinted genes via its own methylated state. Imprinting patterns (i.e., methylation states of ICEs) are formed during gametogenesis with paternal ICEs methylated in sperm and maternal ICEs methylated during oocyte growth.79–81 DNMT3A and DNMT3L are required for the establishment of these imprinting patterns in both cell types and DNMT1 is necessary for maintenance of these imprints.82,83 Histone modifications may play a role in the establishment of these methylation patterns. DNMT3A was shown to recognize H4R2me2 modifications via its plant homeodomain (PHD) motif; the recruitment of DNMT3A leads to de novo methylation of nearby sequences, which was shown in the beta-globulin locus.84 Another experiment has shown that DNMT3L binds to H3, but it is repelled by the H3K4me3 modification.85 This repulsion would prevent de novo methylation of regions marked for poised or active transcription.86,87 The methylation of ICEs results in the transcription of protein-coding genes in cis (e.g., GNAS, IGF2, PWS/AS, KCNQ1). Since DNA methylation is associated with transcriptional silencing, this is an unexpected consequence. DNA methylation at ICEs serves to repress transcriptionally repressive factors, which explains its appearance as an activator. Unmethylated ICEs can form insulators, which prevent mRNA transcription, or unmethylated ICEs lead to the transcription of ncRNAs, which then repress transcription of nearby protein coding genes in a cis fashion.88–90
Imprinting defects, which are seen as the improper establishment of regional DNA methylation haplotypes (i.e., epi-haplotypes), are associated with severe disorders. For example, imprinting defects in chromosome region 15q11.2–q13 are directly linked to Angelman Syndrome (severe developmental delay) and Prader–Willi Syndrome (severe hypotonia).91 Imprinting defects within the imprinted GNAS1 gene lead to pseudohypoparathyroidism, which causes electrolyte imbalances and low-levels of calcium in the blood.92,93 Loss of imprinting at the H19|IGF2 region leads to Beckwith–Wiedemann syndrome (large body size, embryonal tumors, and visceromegaly).94 Allele-specific DNA methylation aberrations also extend beyond imprinted genes. Experiments have shown the expression of the X-linked gene MeCP2 is tightly regulated in vivo. Mutations in this gene cause Rett Syndrome, while a 2X over expression of the gene leads to neurological disorders.95 Severe phenotypes result from deviations from expected ASM patterns, which demonstrate the importance of a tightly regulated methylation regulatory network.
General Allele-Specific Methylation
Beyond imprinted genes whose methylation patterns are determined during gametogenesis, there are additional ASM patterns in mouse and human. A human methylation-sensitive single nucleotide polymorphism (SNP) analysis involving 12 samples covering 6 tissues revealed 16 ASM candidates.56 The authors found that 12 of those sites strongly associated with nearby SNPs. Unlike in imprinted genes, where methylation patterns are determined by parent-of-origin, these ASM sites were dependent on sequence. The authors labeled these ASM regions as epi-haplotypes. Further studying ASM in human, a recent targeted genome survey of CpG islands in chromosome 12 and chromosome 20 across 16 cell lines found that many CpGs with methylation frequencies between 0.25 and 0.75 (‘fuzzily methylated regions’).38,39 Using SNPs to separate alleles in order to explore areas of fuzzy methylation, the authors found between 23 and 37% of heterozygous SNPs showed significant ASM behavior. Due to the high CpG content of the targeted regions, many SNPs overlapped with CpG sites, and many of the ASM examples were due to sequence differences between the alleles (Figures 2c and d). For example, a G/A heterozygous SNP at the second position of a CpG dinucleotide would appear as a CG sequence on one allele and a CA on another allele. The CA sequence would then be completely unmethylated based solely on its sequence. Additional examples of ASM without SNPs at a CpGs were also found. Interestingly, different ASM scales were detected as some regions contained only a single CpG with ASM, while other regions showed ASM spanning several CpGs. A comparison across cell lines revealed that only 6% of ASM behavior was conserved across cells that contained the same heterozygous SNP. Additional experiments in mouse have found hundreds of candidate ASM regions genome-wide.96 Examining 18 of those sites in detail, the authors found 15 of them showed the same methylation levels in the male germ line and were only differentially methylated in somatic cells. Unlike imprinting, the methylation states of these 15 sites are not determined until after fertilization, which means a mechanism separate than imprinting is available for establishing ASM. Many of the ASM regions in this study were associated with sequence variants, which demonstrate the likely role of sequence specific factors in regulating ASM in mouse.97–99 Another genome-wide analysis showed that 2704 SNPs displayed ASM and 90.3% of them were associated with cis-acting ASM.100 ASM flipping has also been observed. ASM flipping describes a SNP region, where allele A is more methylated than allele B in one cell line and allele B is more methylated than allele A in another cell line. Instances of ASM flipping were found across cell line families in our targeted bisulfite sequencing study.39 These studies validate the presence of cis-dependent ASM in various cell lines in both human and mouse. In the mammalian genome, ASM occurs outside of imprinted regions, at different scales, and may be regulated by cis-sequences.
Recent experiments have shown that cell line clonality can lead to biological artifacts and diminishes the in vivo applicability of results based on clonal cell lines. A study found that about 20% of tested lymphoblastoid cell lines were pauciclonal (consisting of only a few clones) or monoclonal.101 In cell lines where only a few clones are present, sensitive allele-specific studies may not reflect the whole cell population in vivo. Such clonal cell lines are likely to exhibit artifacts like random monoallelic expression, which mask the biological signal found in the in vivo cell population. Affected studies include gene expression and methylation studies. Another study found that many traits (e.g., RNA transcript levels, drug responses) are highly variable across lymphoblastoid cell lines and that gene expression is better explained by artifacts (e.g., Epstein-Barr virus (EBV) copy numbers and growth rate) than by genetic variants.102 However, another group that investigated multiple individual-specific cell types showed that clonality did not significantly affect their ASE results.103 Given these findings, experimentalists should account for the clonality of their investigated cell lines when evaluating results, especially in allele-specific studies.
Linked with ASM in many cases, ASE has been observed to occur randomly in clonal cell lines. Experiments comparing the ASE behavior of clonal cell lines derived from B-lymphoblastoid cells revealed the presence of a class of genes whose ASE behavior was random.104 This class describes genes that are expressed on the paternal, maternal, or both alleles across clones. Such genes are spread throughout the genome and do not cluster together in enriched locations. Many of the genes (80%) that showed monoallelic expression in at least one clonal cell line showed biallelic expression in other clonal lines. This demonstrates that these genes can be expressed from either allele or both simultaneously. Overall 5–10% of human autosomal genes were found to be randomly monoallelically expressed. Additional studies need to be performed to demonstrate the prevalence of random monoallelic expression outside of the B-lymphoblastoid cell lines investigated in this paper. Connecting DNA methylation to ASE remains an interesting challenge but methylation studies have the potential to explain many of the recently observed ASE phenomena, including random monoallelic expression.
METHYLATION PATTERN ERASURE: iPS AND PGC DYNAMICS
Reprogramming involves the activation of a small set of transcription factors within a differentiated cell, which transforms the cell into an ES-like state.105–108 The ability to reprogram differentiated cells into a pluripotent state raises hopes for improved transplantation and disease therapies. An issue with transforming cells into iPS cell lines is the low transformation efficiency (<0.1%). The activation of certain transcription factors during transformation may bias the iPS cell toward certain differentiated cell types or lead to tumorigenesis.106,109 In order for iPS-based therapies to become widespread, iPS transformation efficiency and safety must be improved.108,110–115
Methylation and iPS Reprogramming
During differentiation, which is reversed during iPS transformation, massive changes in methylation occur genome-wide. Genome-wide methylome constructions of a fibroblast, IMR90, and an ES cell, H1, have revealed that many regions of the IMR90 genome are methylated at much lower frequency than in H11 (Figure 3a). However, the authors found 491 regions where IMR90 was more methylated than in H1 (DMRs). The 139 genes associated with these DMRs showed higher expression in H1 relative to IMR90 and 113 genes showed lower expression. The majority of these genes had DMRs within 2 kb upstream from the transcription start site (TSS) or 5′ untranslated region. The H1 cell line also showed non-CpG methylation (CHH and CHG) throughout the genome while only CpG methylation was present in IMR90. Relating the methylation signatures of iPS cells to fibroblasts and ES cells, a targeted bisulfite study showed that the methylation signatures of human reprogrammed cells from two fibroblast cell lines clustered closely to ES cells and were distinct from their respective untransformed cell lines38 (Figure 3b). However, the iPS cell lines were still distinguishable from ES cell lines, showing that differences between the iPS cell lines and ES cell lines exist. This difference was also found in another study where iPS cells had regions with methylation signatures that were dissimilar to both ES cells and their respective untransformed cell lines.41 These studies show that there are significant methylation differences between ES and differentiated cells. Successful iPS transformation involves massive methylation changes in order for a differentiated cell to arrive at a pluripotent state.

FIGURE 3 | Methylation frequency differences between differentiated and pluripotent cell lines. (a) This figure shows the methylation status of H1 (ES cell) and IMR90 (fibroblast cell) across a 3 kb region of chromosome 6. Each vertical bar represents a CpG site whose color is dependent on its methylation level. The arrow indicates the promoter of Pou5F1, which codes for the Oct4 protein. Oct4 is a master regulatory of pluripotency in ES cells. The promoter is unmethylated in H1, where Oct4 is transcribed, and methylated in IMR90, where Oct4 is repressed. Regions upstream of the Pou5F1 promoter show larges swaths of differential methylation. Successfully reprogramming involving large methylation changes in such areas. (b) A detailed view of the DNMT3b TSS site across 13 cell lines. The arrows indicate CpG sites that show cell type specific methylation. The iPS lines have methylation signatures that match the ES cells instead of their pretransformed cell types. PGP_1 is a lymphoblastoid cell line while PGP1_F, IMR90, BJ, and hFib2 are fibroblast cell lines. PGP1_iPS1 was transformed from the PGP1 fibroblast. Hues12, Hues42, and Hues63 are embryonic stem cell lines.
Reprogramming involves DNA demethylation and inhibiting DNA demethylation thus diminishes reprogramming efficiency. A mammalian protein associated with demethylation is activation-induced cytidine deaminase, AID, which is a 5-meC deaminase. AID was first discovered as a necessary component in Class Switch Recombination and high levels of Somatic Hypermutation processes, which take place within B-cells.116–118 Initially thought to be expressed only in immune cells, further research showed that AID was also expressed in pluripotent cells.119 AID’s association with demethylation stems from its conversion of methylated cytosines to thymines.120,121 The converted base is removed via G:T mismatch repair, which, for example, can be performed by Mbd4.119,122,123 Demonstrating that the protein AID is important in iPS reprogramming, a group used mouse ES cells fused with human fibroblasts cells to show that AID regulates the transcription of Oct4 and Nanog.124 The fusion of a mouse ES cell and a human fibroblast cell creates a heterokaryon and this process efficiently produces iPS cells. Seventy percent of heterokaryons expressed high levels of human Oct4, Nanog, and GAPDH (negative control protein) transcripts on the third day after fusion. Noticing that AID transcripts were found in the heterokaryons and that AID was bound to the methylated promoter regions of Oct4 and Nanog in human fibroblasts, the authors used several siRNAs that recognized different regions on the AID transcript to knock down AID in both the mouse ES and human fibroblast cells. These siRNAs were then transfused into both cells lines 24 h before the cells were fused. Oct4 and Nanog expression levels were reduced by at least 80% in the AID-knockout heterokaryons relative to the control and bisulfite sequencing revealed significant methylation at the promoter regions of Oct4 and Nanog relative to the control. The results show that the presence of AID likely leads to demethylation at Oct and Nanog promoters, which results in their transcription. Although the knockdown of AID resulted in a significant inhibition of iPS reprogramming, over expression of AID did not change the efficiency of iPS reprogramming. The discovery of AID’s activity during reprogramming does not exclude the possibility of other factors being involved in DNA demethylation. Additionally, the details of AID’s demethylation mechanism in pluripotent cells are still unknown. Additional research is needed to eliminate these ambiguities and undoubtedly show that AID is a necessary factor in active DNA demethylation. Given these caveats, this experiment demonstrates the role of AID in demethylation is important for the reprogramming of somatic cells into a pluripotent state.
Primordial Germ Cell Methylation Erasure
In additional to affecting reprogramming efficiency, AID has been shown to play a role in DNA demethylation in primordial germ cells (PGCs). The erasure of DNA methylation patterns, including those in imprinted regions and the inactivated Xi, is important for PGCs as it eliminates epimutations and allows a return to pluripotency.125–128 Studies have shown that methylation signatures in imprinted regions, single copy genes, and certain repeats (e.g. LINE1) are significantly demethylated in PGCs between 11 and 13.5 days post coitum.129 A recent study expanded on these past findings and looked at methylation effects caused by AID in various genetic elements within murine PGCs.130 Demethylation in PGCs was found be global and included gene regions, transposons, and repeats; the final epigenetic state of PGCs at 13.5 days post coitum was termed an ‘epigenetic ground state’, where the genome is mostly demethylated and histone marks are mostly absent.126,130–133 Comparing Aid−/− knockout to wild-type mice, the authors found that the AID deficient PGCs were significantly more methylated; there was a sex bias as the female AID-knockout cell lines were more methylated than the male knockout cell lines. Interestingly the promoters between the AID knockout and wild-type mice did not show a significant difference; transposons, introns, and exons were more methylated in the knockout line.
RELATIONSHIPS BETWEEN TRANSCRIPTION FACTORS AND DNA METHYLATION
Although genome-wide methylomes have recently become available for various human cell lines, the reasons why genomic regions are unmethylated or methylated are mostly unclear. For example, the observation that CpG islands in the human genome are mostly unmethylated has not been explained on a genome-wide scale. However, studies are beginning to reveal a deeper relationship between specific transcription factors and local DNA methylation patterns (Table 1). These studies serve to build a foundation that will thoroughly explain the nonrandom nature of DNA methylation. A set of three transcription factors that tightly associate with DNA methylation are discussed below.
TABLE 1 | Summary of Mentioned DNA Methylation-Related Transcription Factors
| Transcription Factor | Methylated Binding Specificity | DNA Methylation-Related Consequence of Transcription Factor Binding |
| Cfp1 | Unmethylated | (1) Binds to unmethylated CpG islands and (2) recruits Set1a and Set1b complexes, which trimethylates histones at H3K4 independently of RNA PolII binding.134–136 |
| CTCF | Unmethylated | (1) Blocks enhancer-promoter interactions by acting as an insulator137,138 and (2) protects genomic regions from nearby DNA methylation encroachment.139–141 |
| MeCP2 | Methylated | Associates genome-wide with methylated CpGs in mouse. Leads to H3 deacetylation, which results in global chromatin structure changes.95 Loss of MeCP2 leads to up-regulation of 2,184 genes and down-regulation of 377 genes in mouse.142 |
| Sp1 | Unmethylated | Protects genomic regions from nearby DNA methylation encroachment.143,144 |
The CCCTC-Binding Factor
CTCF contains an 11 zinc finger DNA-binding domain that is highly conserved in higher eukaryotes.145,146 Although initially reported as a silencer, CTCF is a versatile transcription factor that has been seen to enhance and repress transcription at promoters as well as act as an insulator.137,147–150 It binds to a range of sequences and its zinc fingers allosterically customize according to DNA sequence and nearby cofactors, which confers CTCF’s ability to recognize a wide variety of sequences.145
CTCF plays a critical role in the establishment and maintenance of the imprinted H19|IGF2 region. Directly upstream of the H19 locus is an imprint control element (ICE) that contains four CTCF binding sites. This ICE is necessary for the allele specific expression of both H19 and IGF2 genes. Methylation of this ICE on the paternal allele prevents CTCF binding within the ICE and also suppresses H19 expression via promoter methylation.151–153 CTCF binding on the maternal allele prevents enhancers downstream of H19 from interacting with the promoter region of IGF2. Without CTCF’s insulating function on the paternal allele, enhancers downstream of H19 are able to interact with the IGF2 promoter, which leads to the paternal expression of IGF2. Hypomethylation of the H19 promoter on the maternal allele leads to maternal H19 expression. Recent data has shown that CTCF is an essential factor in the formation of long-range contacts (i.e., loops) between this imprinted locus and distal enhancer regions.138,154 In addition to mediating long-range regulatory interactions, CTCF can protect nearby regions from encroaching DNA methylation. An experiment looking at the retinoblastoma tumor suppressor (Rb) gene found that CTCF binding prevents DNA methylation from spreading into the Rb’s CpG island promoter region.155 The ability of CTCF to protect promoters from repressive DNA methylation was also seen in the c-MYC promoter140 and in the BRCA1 promoter.141 These examples show that CTCF binding depends on the methylation state of its binding sequence, and its binding can mediate long-range regulatory interactions and affect local methylation patterns.
CXXC Finger Protein 1
CXXC finger protein 1 (Cfp1) contains a conserved CXXC domain that is sufficient for specific binding to unmethylated CpG dinucleotides and two PHD fingers.156,157 The yeast analog of Cfp1, Spp1, has been found to bind to the H3K4me3 histone modification, which is associated with poised or active transcription, via PHD finger interactions.158 In addition to recognizing H3K4me3 modifications in yeast, Cfp1 associates with the mammalian Set1a and Set1b methyltransferase complexes,135,136 which are known to trimethylate H3K4.
Using ChIP-Seq technology on mouse brain nuclei, a recent study found that Cfp1 was localized at unmethylated CpG islands.134 CpG islands bound by Cfp1 were found to also exhibit H3K4me3 modifications. Unmethylated CpG islands not occupied by Cfp1 or H3K4me3 where found to align with the repressive histone modification mark H3K27me3.159 To test the direct link between Cfp1 and H3K4me3 modification, the authors inserted promoterless CpG-rich DNA into regions not associated with H3K4me3. The results showed Cfp1 binding peaks at the DNA sequence insertion sites and H3K4me3 was present as these locations. However, there was no sign of RNA Pol II binding. This test showed that the H3K4me3 modification is not a byproduct of transcription since it occurs independently of RNA Pol II binding. Cfp1 is a transcription factor that binds to unmethylated CpG islands and modifies nearby histones in a manner that promotes RNA Pol II recruitment.
MeCP2
While the previous two transcription factors bind to unmethylated sequences, methyl CpG binding protein 2 (MeCP2) recognizes CpG methylated DNA.160,161 MeCP2 is a part of the MBD protein family (MeCP2, MBD1, MBD2, and MBD4), which consists of proteins that share a homologous methylated DNA recognition domain, but contain different transcriptional repression domains.162–164 The mammalian form of MBD3 is an exception, since there is a mutation in its MBD that prevents it from binding to methylated CpGs.165 MeCP2 binding was originally thought to cause transcriptional repression due to its recruitment of histone modifying enzymes, which results in repressive histone marks around regions bound by MeCP2, namely histone deacetylation and trimethylation of H3K9.166–168 MeCP2 is expressed highly only in neurons and its expression level is tightly regulated. Embryonic MeCP2-null mice die at week 12 while mature mice under- or over-expressing MeCP2 (e.g., heterozygous females) exhibit neurological disorders.169–172
The transcriptional role of MeCP2 has recently changed from a transcriptional repressor to a global chromatin remodeler that can lead to the expression and repression of various genes. Since MeCP2 binding leads to traditionally associated transcriptionally repressive histone modification patterns, MeCP2 binding near genes was thought to silence them. However, a recent study showed that mice over-expressing MeCP2 led to a significant number of up regulated genes relative to MeCP2-null mice (2184 genes up regulated while 377 genes down regulated).142 These authors found that a global transcriptional activator CREB1 and MeCP2 co-occupied promoters of many activated genes, including the Creb1 promoter, and CREB1 was also found to copurify with MeCP2. These findings are inconsistent with the model of MeCP2 as a transcriptional repressor. Further changing the view of MeCP2’s role as a repressor, another study performed on mouse brain tissue revealed MeCP2 binding correlates with DNA methylation levels genome-wide. MeCP2 is a protein highly expressed in neuronal cells and its concentration in neurons is similar to that of nucleosomes.95 Using ChIP-Seq to enrich for sequences bound to MeCP2, the authors found that 56% of the mouse genome showed some MeCP2 binding activity. The intensity of the MeCP2 signal increased as DNA methylation density increased. MeCP2 binding was not found at unmethylated CpG islands, suggesting a genome-wide methylated DNA binding preference for MeCP2. To test the relationship between H3 acetylation and MeCP2 abundance, the level of H3 acetylation was measured between wild-type and MeCP2-null neurons. There was a 2.6-fold increase in H3 acetylation in the MeCP2-null neurons relative to the wild-type. As a control, wild-type and MeCP2-null glial cells were also examined, but no significant differences in H3 acetylation were found. The MeCP2-null brain cell lines also showed a 1.6-fold increase in transcription of repetitive elements, while no expression change was seen in Actb, c-Myc, or tyrosine hydroxylase genes. These authors present MeCP2 as a protein that binds to methylated DNA on a global scale. In accordance with previous experiments, MeCP2 does reduce H3 acetylation levels and inhibits transcription of repetitive elements. Due to the global scale of histone deacetylation in the presence of MeCP2, MeCP2’s association with deacetylation, which is traditionally associated with silencing transcription, and activation of gene transcription are not necessarily mutually exclusive. The consequences of global histone modification changes are unknown and a rigorous study into this subject may bridge these two seemingly opposing characteristics of MeCP2.
Transcription Factors Regulate and Are Regulated by DNA Methylation
Experiments focused on specific transcription factors have shown the dependence of DNA methylation states on transcription factor binding. However, the list of transcription factors affected by methylation is short and the relationships between DNA methylation and most transcription factors are still unclear. A recent motif based study looked at CpG island sequences that are resistant to de novo methylation in colorectal and leukemia cells. Using motif analysis tools, the authors found a set of motifs that are strongly associated with de novo methylation resistance.173 The most significant motifs are YY1, Sp1, and NRF1. Sp1 has been known to protect CpG islands from methylation.143,144 YY1 recruits the Polycomb Repressive Complex 2, PRC2, which is known for transcriptional silencing via the H3K27me3 histone modification.174 PRC2 may also act in a context-specific manner as an allele-specific antagonist to DNA methylation.175 Elucidating the causes and consequences of DNA methylation will yield a better understanding of cell differentiation, reprogramming to pluripotency, and cancer development since these processes involve massive genome-wide methylation changes.
HYDROXYMETHYLCYTOSINE: A NEW CYTOSINE MODIFICATION
Complementing AID’s demethylation activities via deamination and G:T mismatch repair, a new family of mammalian proteins have been reported to convert methylated cytosine (5meC) bases into hydroxymethylcytosine (5hmC) in vivo176,177 (Figure 4). The first study identified an unknown base that started appearing on Thin Layer Chromatography plates.176 The authors found that 5hmC comigrated with this unknown spot and mass spectrometry later confirmed the presence of this compound as 5hmC. This new cytosine modification was found the nuclei of Purkinje neurons and granule cells. Hmc5 was found to make up 0.59% of all bases in Purkinje DNA and 0.23% in granule cell nuclei. A seemingly insignificant fraction of DNA exhibits this modification but relative to CpG, which makes up about 1% of all bases, the abundance of 5hmC is indeed significant. Another study searched for proteins responsible for this novel modified base. Trypanosome proteins JPB1 and JPB2 are known to hydroxylate and glucosylate the methyl group in a thymine, which results in b-d-glucosyl hydroxymethyluracil.178 Using the predicted oxygenase domain in those proteins to find proteins with similar functions in human, this study found that the predicated trypanosome oxygenase domain shared significant homology with the human proteins TET1, TET2, and TET3. To test for TET1’s activity, the authors transfected hemagglutinin-tagged TET1 into embryonic kidney cells (HEK 293). Kidney cells that showed an increasingly strong signal for hemagglutinin (HA) also showed a decreasing signal for methylated cytosine (5meC) relative to a mock control. Using methylation-sensitive restriction enzyme techniques, the authors discovered a base of unknown identity. Using mass spectrometry and comparing with the fragmentation pattern of 5hmC, the authors found that the unknown base was hmC. Furthermore, the authors found that 5hmC made up 4–6% of all cytosine species at MspI cleavage sites in mouse ES cells while 5meC made up 55–60%. Knockdown of TET1 via RNAi led to a 40% decrease in 5hmC levels. The continued presence of 5hmC in the absence of TET1 was attributed to the presence of additional proteins, such as TET2 and TET3. Finally, the induced differentiation of mouse ES cells by removal of LIF for 5 days led to an 80% TET1 transcript decline and about a 40% drop in 5hmC levels. Protocols for large-scale identification of 5hmC have yet been presented, but a recent study evaluated the ability of current methylation detection techniques to detect 5meC, 5hmC, or both. 5hmC appears to be resistant to bisulfite and bisulfite sequencing results thusly do not distinguish between 5meC and 5hmC. Polymerase chain reaction (PCR) appears to amplify 5hmC and meC sequences with similar efficiency. The monoclonal antibody used in MeDIP experiments,179–181 however, is 5meC specific. The proteins MBD2b, MBD1, MBD4, and MeCP2 also bind 5meC specifically.162,182–184 These studies show the presence of another type of modified cytosine in vivo. Bilsulfite sequencing cannot discern between the cytosine types but MeDIP and certain protein complexes can. Recent evidence showed that single molecule real time sequencing technology (SMRT) discriminates between the two types of cytosine modifications, too. The presence of 5meC and 5hmC affects polymerase kinetics in SMRT and this can be exploited to differentiate between the two modifications at a base pair resolution.185
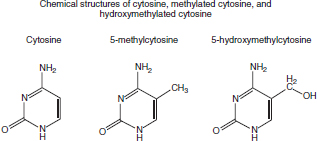
FIGURE 4 | Structures of in vivo cytosine modifications found in humans. This figure shows the structure of cytosine and its two modified forms found in the human genome.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


