Jared R. Brosch, Martin R. Farlow
Alzheimer Disease
Alzheimer disease (AD) is the most common cause for dementia affecting older adults. The illness was first described by Alois Alzheimer in 1906 in a 51-year-old woman with well-described features of dementia. After death, her brain was examined and found to have numerous cortical plaques and tangles, characteristic of the illness.1 The disease was thought to be rare for 6 decades until its clinical and neuropathologic features were recognized to be largely identical to those occurring in older dementia patients.2
The illness is the most frequent cause of dementia in the United States, currently afflicting as many as 5.2 million mostly older adults.3 The incidence doubles every 6 years after the age of 50 years. As the over-80 population has rapidly grown, with improved health care leading to longer survival, it is estimated that the prevalence of AD in the United States by 2050 will increase to 13.8 million Americans, with 7.0 million older than 85 years.4 This disabling disease is the most common cause of neurodegeneration and is the major condition leading to nursing home placement. The economic costs are huge, with direct health care, nursing home costs, and caregiving costs at home globally totaling greater than $315 billion.5 Only symptomatic therapies are currently available, but no known therapy delays or halts disease progression. If a drug or lifestyle alteration (e.g., diet, exercise) could be found that delayed functional deterioration by as little as 1 to 2 years, it would substantially reduce the costs to families and society.6
Pathology and Mechanisms of Disease
In AD, the illness begins with atrophy in the entorhinal cortex and hippocampus and, as clinical symptoms worsen, there is spread to most areas of the cortex, except for the occipital lobes.7
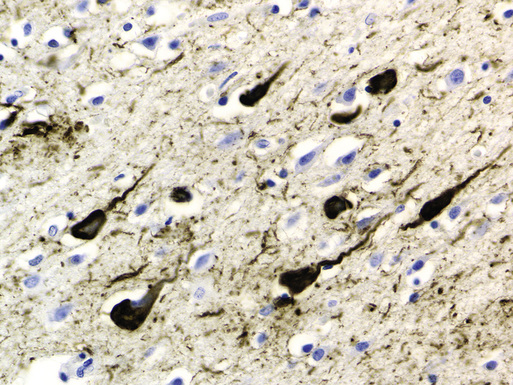
Microscopic examination of cortical sections from the brain of a patient with AD reveals global loss of neurons and increased numbers of extracellular amyloid plaques.8 The amyloid plaques are diffuse and possibly benign, because similar deposits may be found in normal older adults, and compact, often associated with neuritic changes, possibly caused by effects of the deposited amyloid or by smaller oligomers of the β-amyloid protein on surrounding dendrites and axons (Figure 52-1). β-Amyloid proteins are also frequently found deposited diffusely in cerebral blood vessel walls. In addition to β-amyloid plaques, intracellular lacy fibrillar accumulations of material are present in neurons, called neurofibrillary tangles (NFTs; Figure 52-2).9 NFTs are composed of paired helical filaments largely consisting of abnormally hyperphosphorylated tau. These are the defining features originally described by Alzheimer 102 years ago.
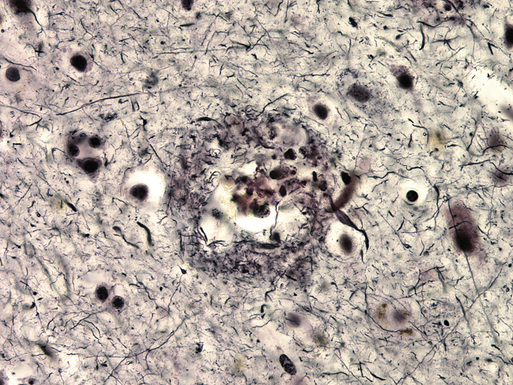
The amyloid plaques are made of β-amyloid proteins of various lengths, from 39 to 42 amino acids long.10 The β-amyloid 1-40 amino acid type is in plasma and cerebrospinal fluid (CSF), but the 1-42 form comprises the major component of the cores of these amyloid plaques. The β-amyloid proteins are derived from a larger transmembrane protein that is widely present in the brain, the amyloid precursor protein (APP). They are cleaved from APP by the β-secretase and γ-secretase enzymes, which have been characterized and are active targets for drug therapy.11
Mutations have been found to be associated with familial AD, as will be described later in this chapter, in the genes coding for the β-amyloid proteins and for the presenilins, which likely are a component of γ-secretase.12 Studies of the effects of these mutations in cell lines and transgenic animals that develop amyloid deposits and plaques in their brains, similar to patients with AD, have provided insights into how these proteins normally function and how, when disturbed by mutation, their functions are altered, leading to dementia. Those findings should provide insights into sporadic AD and, using these models, several drugs have been developed to reduce production of the β-amyloid proteins or speed their metabolism and clearance. It is hoped that these drugs will also be effective for sporadic AD.
Interestingly, although considerable evidence has suggested that β-amyloid and its toxic effects on the brain may initiate the cascade of pathophysiologic processes that leads to AD, the progression of dementia correlates more closely to numbers of NFTs or number of synapses lost.
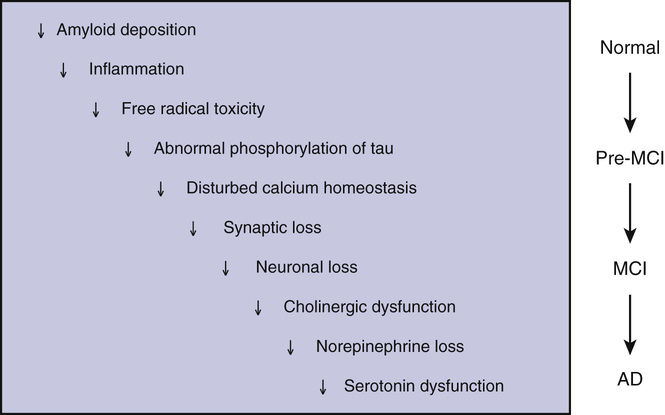
Clearly, a cascade of processes involving different mechanisms, including inflammation and disturbed calcium homeostasis, occurs as AD progresses (Figure 52-3). Neurotransmitter systems are differentially affected. The cholinergic system is susceptible to early dysfunction, and increasing cholinergic deficiency has been correlated with clinical progression. As AD progresses, the glutaminergic, noradrenergic, and serotonergic systems deteriorate, causing further cognitive losses and often behavioral abnormalities. Therapeutic research since the late 1980s has focused on correcting these neurotransmitter deficits, with some modest success and resulting clinical benefits.
Extracellular amyloid plaques and intraneuronal NFTs in the cortex are the cardinal features of AD. Stereotypic spread of the NFTs, as originally described by Braak and Braak (1991),7 defined a basis for neuropathologic criteria for AD staging that is still useful today. NFTs are found in the entorhinal cortex and adjacent areas of the hippocampus. Braak stages I and II typically are not associated with clinical symptoms. In stages III and IV, NFTs have spread to the limbic regions, and cognitive deficits are present. By stages V and VI, NFTs have spread to other regions of the cortex except the occipital cortices, and the severity of the dementia is worse.
Newer criteria assessing plaque and tangle densities have been developed. Khachaturian criteria count numbers of amyloid plaques in a 1-mm2 field of the neocortex, with numbers adjusted for age.13 The Consortium to Establish a Registry for Alzheimer’s Disease criteria count plaques in three designated brain regions using defined stains.14 The Reagan criteria assess plaques and tangle numbers in a highly specified way and appear to have generally high correlation in confirming the diagnosis of AD and assessing disease stage.15
Substantial evidence has suggested that plaques and tangles can be found in great numbers in many older adults with normal cognitive functioning, and that considerable overlap exists in plaque and tangle pathology between very old normals (>90 years) and those in this age range with dementia, reemphasizing the necessity for clinicopathologic correlation in diagnosis.16 Although these pathologic features correlate less well in the oldest old, the presence of neocortical atrophy remains a strong associated factor for Alzheimer dementia at any older age.17
Epidemiology and Genetics
Aging is the biggest risk factor for AD. Rarely, patients in their 20s and 30s have been reported to have AD, but the onset of clinical symptoms is uncommon until the 50s, with yearly probability of incidence rapidly increasing to age 65 to 75 years, when 1% to 5% of the general population is affected. By age 75, the prevalence is as high as 15% in the United States, and by age 85, it has been estimated to afflict 35% to 50% in the oldest old.18 Studies have suggested that the prevalence continues to climb, with most older adults by their 90s showing clinical signs of at least early dementia. However, in these oldest old with dementia, underlying brain pathology may differ little from age-matched normal subjects other than that demented patients have greater neuronal losses, and their rate of disease progression may be relatively slow. Therefore, even though these patients meet current clinical criteria for AD, it is unclear that they really have the same illness and may have a larger contribution from underlying vascular disease.19
The second most common risk factor for AD is family history, with approximately 20% of patients with AD having two or more first-degree relatives affected. The pattern of inheritance is typically autosomal dominant. In families with AD, several genetic mutations have been identified that are causative for the disease. Almost all the known mutations cause presenile forms of the disease. A few dozen families with onset of AD predominantly in their 40s and 50s have mutations in the APP gene, usually in the region of the gene that codes for the β-amyloid proteins. It is thought that these mutations result in abnormal β-amyloid metabolism, with resultant chronically higher levels of the protein leading to AD.20
The other mutations causing early-onset familial disease have been localized to the presenilin-1 (PS-1) gene on chromosome 14 and the presenilin-2 (PS-2) gene on chromosome 1. Again, only a few dozen families have been found in which PS-2 gene mutations are associated with AD, whereas more than 160 different mutations in the PS-1 have been found in several hundred families to cause AD and occasionally other symptoms, such as spasticity, seizures, or ataxia.21,22 The presenilins are part of a complex that acts functionally as γ-secretase.12 As noted, γ-secretase slices APP to produce β-amyloid.
Increased levels of β-amyloid are also found in AD patients with PS-1 or PS-2 mutations.20 These mutations are the most commonly known cause of autosomal dominant familial AD, but they still cause less than 1% of all cases of AD. Even in patients with a family history of onset of dementia in those younger than 65 years, these mutations are causative in less than 10%. An early-onset patient with AD is more likely to have one of these mutations if there is a family history of other affected family members having a similar early age of onset. Other than aid in confirming the diagnosis or risk for AD in a limited number of these relatively rare families, the major importance of these mutations has been to create transgenic animals that have been useful in the following: unraveling disease mechanisms; hastening the development of new drugs and testing procedures; and finding other novel therapeutic approaches, such as targeted immunologic stimulation by β-amyloid vaccination or administering monoclonal antibodies targeted to the β-amyloid proteins to accelerate their degradation.
The major genetic risk factor identified for AD in late-onset, sporadic, and familial patients is the ε4 polymorphism of the apolipoprotein E (ApoE) gene.23 ApoE is one of the major cholesterol- and lipid-carrying proteins in peripheral blood. In the brain and spinal fluid compartments, it is the only significant lipid transport protein and also functions as a transport protein for β-amyloid. It comes as types ε2, ε3, and ε4, with the ε2 allele being relatively uncommon. The ε4 allele is carried by 15% to 20% of the general population and the ε3 allele by almost everyone else. The 1% to 2% of individuals who are homozygous for the ε4 allele have a 50% risk of developing AD by their mid to late 60s. Those who are heterozygous for the ε4 allele have a 50% risk of developing AD by their mid to late 70s.23 Women heterozygous for ε4 are more likely to develop AD at an earlier age than men.24 Individuals who carry only the ε2 or ε3 allele are likely not to develop AD in their 80s or not at all. Some evidence has suggested that inheriting the ε2 allele may be protective, reducing the risk for AD. Inheritance of the ε4 allele confers a similar risk for AD, regardless of whether or not there is a family history.
Determination of the ApoE genotype in a patient with dementia improves diagnostic specificity in the subset of subjects with the ε4 genotype, but it is arguable whether diagnostic accuracy is improved enough to be clinically helpful. If subjects who are homozygous or heterozygous for ε4 and do not develop dementia in their at-risk age range, current evidence suggests that their future risk of dementia is no greater than that of an age-matched, non-ε4 carrier in the general population for developing AD. Therefore, presymptomatic ApoE ε4 genotyping for future risk for AD is currently not recommended. Interestingly, ApoE ε4 patients have been found to recover less completely from head trauma, so a more prominent history of head trauma may be a pseudomarker for carrying the ApoE ε4 polymorphism.25 It has been found that in individuals with mild cognitive impairment (MCI), the ApoE ε4 genotype is associated with a significantly greater risk for conversion to AD,26 and ε4-carrying patients with MCI were more responsive to donepezil therapy.27 Clinical disease progression after conversion to AD is not influenced by the ApoE genotype, so the ApoE ε4 protein may be more influential in the earlier biologic stages of the illness. However, once disease spreads and the cascade of destructive pathophysiologic processes begins, the ApoE genotype no longer influences the rate of progression. Considerable evidence in late-onset AD has suggested that there are multiple genetic polymorphisms that may play a role in increasing the risk for some patients to develop the illness.28,29
Other factors that influence risk for AD are gender and education. Women are at greater risk for AD, even with adjustment for their longer survival to an older age. In the past, this greater risk for AD has been suggested by several epidemiologic studies to be due to postmenopausal estrogen deficiency, and estrogen replacement was believed to be primary prevention for AD. However, the Women’s Health Initiative Memory Study of estrogen in older women has shown that estrogen replacement may increase, rather than decrease, the risk for dementia.30,31
In several studies, increased education has been associated with reduced overall risk for AD and/or later onset of the illness. It has been hypothesized that better educated individuals have a greater cognitive reserve, so the underlying biologic process of disease must progress further for clinical symptoms to develop.32,33 However, the protective effects of cognitive training and mental exercises have not demonstrated convincing benefits in delaying disease onset. Interestingly, several studies have suggested that physical exercise may be protective and decrease brain atrophy and/or delay disease progression.34 A large, multicenter, randomized controlled trial investigating exercise in AD is currently underway, and results will be available after 2018.35
Epidemiologic studies and small pilot trials have suggested that nonsteroidal antiinflammatory drugs (NSAIDs) prevent or delay progression in AD.36 However, several large double-blind, placebo-controlled trials have indicated no significant reduction in risk for AD with NSAIDs but have shown a significantly increased risk for gastrointestinal symptoms, such as hemorrhage, and cardiovascular disease, including stroke.37,38
A number of studies and trials have suggested that the metabolic syndrome increases the risk for AD.39 Specifically the main features of the metabolic syndrome-diabetes mellitus or insulin resistance, high cholesterol level, hypertension, and obesity-are all suggested risk factors for AD. Several prospective studies have been investigating whether improving or treating these factors individually will reduce conversion of normal or MCI individuals to AD and/or delay progression of AD in subjects already affected. It remains to be determined whether these approaches, such as treating hypertension and treating insulin resistance with glitazones and statins to reduce cholesterol, are effective.
A growing body of evidence has found an association between physical frailty and AD incidence. A study of 823 patients demonstrated that each unit change in baseline frailty score led to a more than 9% increased risk of developing AD.40 Frailty in this study was a composite score involving grip strength, body mass index, walking speed, and assessment of fatigue. An autopsy study also demonstrated an increased risk of AD pathology, with or without the presence of clinical dementia, in those with higher frailty scores.41 Frailty is also associated with mild cognitive impairment and has an impact on quality-of-life scores in those with AD.42,43
Epidemiology and Genetics
Aging is the biggest risk factor for AD. Rarely, patients in their 20s and 30s have been reported to have AD, but the onset of clinical symptoms is uncommon until the 50s, with yearly probability of incidence rapidly increasing to age 65 to 75 years, when 1% to 5% of the general population is affected. By age 75-85 the prevalence is otherwise as high as 18% in the United States, and over age 85, it has been estimated to afflict 33% to 42%.4 Studies have suggested that the prevalence continues to climb, with most older adults by their 90s showing clinical signs of at least early dementia. However, in these oldest old with dementia, underlying brain pathology may differ little from age-matched normal subjects other than that demented patients have greater neuronal losses, and their rate of disease progression may be relatively slow. Therefore, even though these patients meet current clinical criteria for AD, it is unclear that they really have the same illness and may have a larger contribution from underlying vascular disease.19
The second most common risk factor for AD is family history, with approximately 20% of patients with AD having two or more first-degree relatives affected. The pattern of inheritance is typically autosomal dominant. In families with AD, several genetic mutations have been identified that are causative for the disease. Almost all the known mutations cause presenile forms of the disease. A few dozen families with onset of AD predominantly in their 40s and 50s have mutations in the APP gene, usually in the region of the gene that codes for the β-amyloid proteins. It is thought that these mutations result in abnormal β-amyloid metabolism, with resultant chronically higher levels of the protein leading to AD.20
The other mutations causing early-onset familial disease have been localized to the presenilin-1 (PS-1) gene on chromosome 14 and the presenilin-2 (PS-2) gene on chromosome 1. Again, only a few dozen families have been found in which PS-2 gene mutations are associated with AD, whereas more than 160 different mutations in the PS-1 have been found in several hundred families to cause AD and occasionally other symptoms, such as spasticity, seizures, or ataxia.21,22 The presenilins are part of a complex that acts functionally as γ-secretase.12 As noted, γ-secretase slices APP to produce β-amyloid.
Increased levels of β-amyloid are also found in AD patients with PS-1 or PS-2 mutations.20 These mutations are the most commonly known cause of autosomal dominant familial AD, but they still cause less than 1% of all cases of AD. Even in patients with a family history of onset of dementia in those younger than 65 years, these mutations are causative in less than 10%. An early-onset patient with AD is more likely to have one of these mutations if there is a family history of other affected family members having a similar early age of onset. Other than aid in confirming the diagnosis or risk for AD in a limited number of these relatively rare families, the major importance of these mutations has been to create transgenic animals that have been useful in the following: unraveling disease mechanisms; hastening the development of new drugs and testing procedures; and finding other novel therapeutic approaches, such as targeted immunologic stimulation by β-amyloid vaccination or administering monoclonal antibodies targeted to the β-amyloid proteins to accelerate their degradation.
The major genetic risk factor identified for AD in late-onset, sporadic, and familial patients is the ε4 polymorphism of the apolipoprotein E (ApoE) gene.23 ApoE is one of the major cholesterol- and lipid-carrying proteins in peripheral blood. In the brain and spinal fluid compartments, it is the only significant lipid transport protein and also functions as a transport protein for β-amyloid. It comes as types ε2, ε3, and ε4, with the ε2 allele being relatively uncommon. The ε4 allele is carried by 15% to 20% of the general population and the ε3 allele by almost everyone else. The 1% to 2% of individuals who are homozygous for the ε4 allele have a 50% risk of developing AD by their mid to late 60s. Those who are heterozygous for the ε4 allele have a 50% risk of developing AD by their mid to late 70s.23 Women heterozygous for ε4 are more likely to develop AD at an earlier age than men.24 Individuals who carry only the ε2 or ε3 allele are likely not to develop AD in their 80s or not at all. Some evidence has suggested that inheriting the ε2 allele may be protective, reducing the risk for AD. Inheritance of the ε4 allele confers a similar risk for AD, regardless of whether or not there is a family history.
Determination of the ApoE genotype in a patient with dementia improves diagnostic specificity in the subset of subjects with the ε4 genotype, but it is arguable whether diagnostic accuracy is improved enough to be clinically helpful. If subjects who are homozygous or heterozygous for ε4 and do not develop dementia in their at-risk age range, current evidence suggests that their future risk of dementia is no greater than that of an age-matched, non-ε4 carrier in the general population for developing AD. Therefore, presymptomatic ApoE ε4 genotyping for future risk for AD is currently not recommended. Interestingly, ApoE ε4 patients have been found to recover less completely from head trauma, so a more prominent history of head trauma may be a pseudomarker for carrying the ApoE ε4 polymorphism.25 Recently, it has been found that in individuals with mild cognitive impairment (MCI), the ApoE ε4 genotype is associated with a significantly greater risk for conversion to AD26 and that ε4-carrying patients with MCI were more responsive to donepezil therapy.27 Clinical disease progression after conversion to AD is not influenced by the ApoE genotype, so the ApoE ε4 protein may be more influential in the earlier biologic stages of the illness. However, once disease spreads and the cascade of destructive pathophysiologic processes begins, the ApoE genotype no longer influences rate of progression. Considerable evidence in late-onset AD has suggested that there are multiple genetic polymorphisms that may play a role in increasing the risk for some patients to develop the illness.28,29
Other factors that influence risk for AD are gender and education. Women are at greater risk for AD, even with adjustment for their longer survival to an older age. In the past, this greater risk for AD has been suggested by several epidemiologic studies to be due to postmenopausal estrogen deficiency, and estrogen replacement was believed to be primary prevention for AD. However, the Women’s Health Initiative Memory Study of estrogen in older women has shown that estrogen replacement may increase, rather than decrease, the risk for dementia.30,31
In several studies, increased education has been associated with reduced overall risk for AD and/or later onset of the illness. It has been hypothesized that better educated individuals have a greater cognitive reserve, so the underlying biologic process of disease must progress further for clinical symptoms to develop.32,33 However, the protective effects of cognitive training and mental exercises have not demonstrated convincing benefits in delaying disease onset. Interestingly, several studies have suggested that physical exercise may be protective and decrease brain atrophy and/or delay disease progression.34 A large, multicenter, randomized controlled trial investigating exercise in AD is currently underway, and results will be available after 2018.35
Epidemiologic studies and small pilot trials have suggested that nonsteroidal antiinflammatory drugs (NSAIDs) prevent or delay progression in AD.36 However, several large double-blind, placebo-controlled trials have indicated no significant reduction in risk for AD with NSAIDs but have shown a significantly increased risk for gastrointestinal symptoms, such as hemorrhage, and cardiovascular disease, including stroke.37,38
A number of studies and trials have suggested that the metabolic syndrome increases the risk for AD.39 Specifically the main features of the metabolic syndrome—diabetes mellitus or insulin resistance, high cholesterol level, hypertension, and obesity—are all suggested risk factors for AD. Several prospective studies have been investigating whether improving or treating these factors individually will reduce conversion of normal or MCI individuals to AD and/or delay progression of AD in subjects already affected. It remains to be determined whether these approaches, such as treating hypertension and treating insulin resistance with glitazones and statins to reduce cholesterol, are effective.
A growing body of evidence has found an association between physical frailty and AD incidence. A study of 823 patients demonstrated that each unit change in baseline frailty score led to a more than 9% increased risk of developing AD.40 Frailty in this study was a composite score involving grip strength, body mass index, walking speed, and assessment of fatigue. An autopsy study also demonstrated an increased risk of AD pathology, with or without the presence of clinical dementia, in those with higher frailty scores.41 Frailty is also associated with mild cognitive impairment and has an impact on quality of life scores in those with AD.42,43
Clinical Diagnostic Criteria
Criteria for the clinical diagnosis of AD have been developed and are widely used to diagnose this form of dementia. These are the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-V),44 and consensus criteria developed by the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA),45 which were revised in 2011 by the National Institute on Aging and the Alzheimer’s Association (NIA-AA).46–48
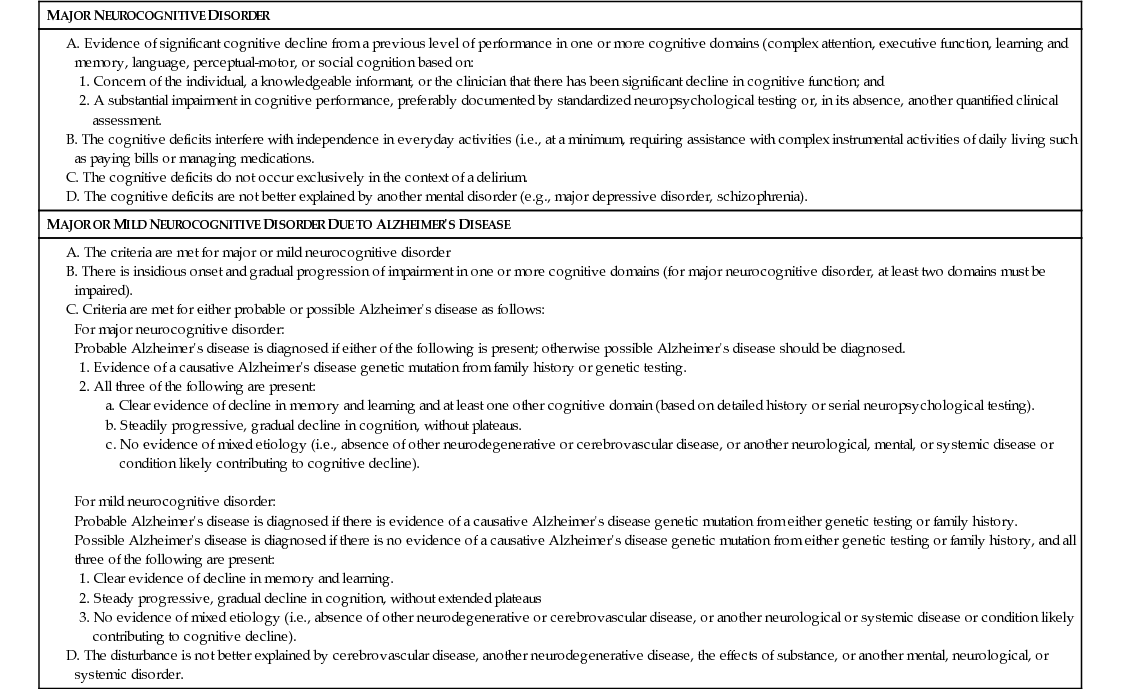
The DSM-V criteria are more general, essentially requiring insidious and progressive memory and cognitive impairments when other potential causes have been excluded (Table 52-1). In the DSM-V criteria, dementia is regarded as a major neurocognitive disorder, and specific criteria for AD and others are detailed. Genetic testing has been included in the DSM-V criteria, but biomarker and imaging results were not incorporated.
TABLE 52-1
DSM-V Criteria for Dementia
The NIA-AA criteria retain much of the original NINCDS-ADRDA criteria. One change is the incorporation of biomarker data into the diagnosis of preclinical dementia, MCI, and AD. Although preclinical dementia and MCI are considered separately, it is recognized that they are all on a spectrum of disease based on advancements in histopathologic understanding of AD. Further refinement to the staging and subtypes of AD are other notable changes in the updated criteria. The new criteria delineate between criteria to be used clinically and criteria that help more clearly define research in AD.
In the revised NIA-AA criteria, preclinical disease is based on biomarker data and is intended primarily for research purposes. There are three defined stages of preclinical disease that involve a combination of imaging and CSF data. Subtle neuropsychological changes, which do not meet criteria for MCI, are included in the final stage of preclinical disease. MCI criteria are primarily clinical; however, additional biomarker data can be used to supplement the diagnosis. The likelihood that MCI is caused by AD has also been proposed to allow further refinement of the criteria. For diagnosis of AD dementia, a set of core clinical criteria must be met (Table 52-2). These incorporate the symptoms of the less common presentation of AD, including logopenic primary progressive aphasia and posterior cortical atrophy phenotypes. Definitions of “possible AD” and “probable AD” diagnoses, as well as the incorporation of optional biomarkers into these diagnoses, are included. Finally, criteria for dementia unlikely to be due to AD and pathophysiologically proven AD dementia are defined.
TABLE 52-2
National Institute on Aging–Alzheimer’s Association Criteria for Alzheimer Disease (AD)








