Currently, six parenteral iron preparations are available, although carboxymaltose (Injectafer) is not licensed in the US (Table 18.1). No randomized trials have compared the safety and efficacy of these agents to one another. However administration of intravenous iron is known to enhance the erythropoietic response to anemia five-fold in normal subjects, which approximates the maximum rate of red cell production possible without substantial expansion of the functional bone marrow space (Hillman and Henderson 1969). Much of the infused iron will be sequestered eventually in the reticuloendothelial system and may not be easily mobilized subsequently for erythropoiesis. Studies of total dose replacement with radiolabelled iron dextran for patients with iron deficiency indicate that 50% of IV iron will be incorporated into hemoglobin within 3 to 4 weeks (Wood et al. 1968).
Not-So-Simple Iron Deficiency
Whereas most patients who are anemic and iron deficient respond adequately to oral iron, for some patients, (e.g. anemic women post-partum, women who have ongoing blood loss from severe menorrhagia, patients who suffer gastrointestinal blood loss, or are intolerant or non-compliant), intravenous iron administered as a single total dose replacement or in divided doses can raise the hemoglobin level faster than can oral iron and can restore bone marrow iron stores (Van Wyck et al. 2007, 2009). Because the safety and efficacy of the two different methods of intravenous iron administration are similar, convenience should determine whether repeated boluses or total-dose infusion is used. If boluses are used, doses should not exceed 400 mg per visit. Doses of 100–200 mg iron are administered as a 2-min intravenous push or diluted in normal saline as a 30-min infusion. Doses for total-dose infusion should be diluted in 500 ml normal saline, infused over 3–4 h after a test dose of the diluted solution.
For total-dose infusion, the low-molecular-weight iron dextran is the preferred preparation, because the iron salts cause dose-dependent gastrointestinal and vasoactive reactions at doses above 200–400 mg (Chandler et al. 2001). The dose, in mg iron, is calculated by: Total iron requirement (cumulative dose) in mgs: Patient weight in kg × (15 − current Hb g/dL) × 2.4 + 500. An intravenous test dose of 25 mg of the diluted solution is required. Administration of corticosteroids before the test and after the infusion decreases the incidence of myalgias and arthralgias (Auerbach et al. 1998). For patients receiving cyclical therapies such as cancer chemotherapy and haemodialysis, short 100–400 mg infusions can be used. No serious adverse events have been observed after more than 20 000 doses of low-molecular-weight iron dextran (Auerbach et al. 2007).
Chronic Renal Failure
Chronic renal failure is an inflammatory condition that results in inhibition of iron absorption from the small bowel and iron recycling from macrophages. Inhibition of iron release can result in both relative iron deficiency and anemia. Infusion of intravenous iron dextran lowers erythropoietin requirement in hemodialysis patients (Panesar and Agarwal 2002). Trials of iron to date have all used both erythropoietic and iron agents. No trial using erythropoietin alone, iron alone, or non-iron adjuvants alone has been designed to investigate the risks and benefits of achieving a similar Hb endpoint. Patients with chronic renal disease not on dialysis who are receiving erythropoietic agents may also benefit from intravenous iron; in this instance the total dose regimen may be more convenient than administering iron during repeated visits. The use of intravenous iron in dialysis patients suggests that most serious adverse events are associated with HMW iron dextran product and are rare (<1:200 000) with low-molecular-weight iron dextran or the other preparations (Chertow et al. 2006). Adverse event rates might be somewhat higher in patients with inflammatory diseases in which immune-mediated drug reactions may be observed more commonly than in dialysis patients. An analysis of adverse events reported to the FDA suggest that more events are seen with HMW iron dextran, but these data fall far short of controlled trials (Wysowski et al. 2010).
Malignancy
Patients with cancer undergoing chemotherapy, many of whom have anemia related in part to poor iron mobilization appear to respond better to erythropoietic agents when parenteral iron is administered, independent of stainable marrow haemosiderin (Auerbach et al. 2004; Hedenus et al. 2007).
Randomized trials have compared the efficacy of oral and intravenous iron in anaemic patients with cancer receiving recombinant erythropoietin. Oral iron with recombinant erythropoietin was not significantly better than recombinant erythropoietin alone, but intravenous iron with recombinant erythropoietin resulted in a significantly better erythropoietic response similar to the studies of patients on dialysis (Auerbach et al. 2004; Henry et al. 2007).
Concern still exists about the long-term effects of intravenous iron on clinical outcomes. Although intravenous iron treatment is standard of care for haemodialysis patients to improve haemoglobin concentrations and reduce erythropoietin requirements, no iron intervention trials have been sufficiently powered to assess safety and long-term clinical outcomes (Sullivan 2004; Zacharski et al. 2007; National Kidney Foundation 2006). Infection is a major cause of mortality and morbidity in dialysis and cancer patients; certain ‘siderophilic’ microorganisms use iron as a metabolic cofactor and can thus become either more frequent, or more virulent as a result of iron use (Melby et al. 1982). Furthermore, intravenous iron increases oxidative stress and impairs endothelial function, both of which could theoretically increase the risk of cardiovascular events in CKD patients (Brewster 2006). The independent contribution of this intervention to cardiovascular events and mortality in such patients warrants further investigation.
Haematopoietic Growth Factors
Haemopoietic growth factors produced by recombinant technology have enjoyed increasing use in clinical medicine, and in many instances their administration has either complemented or replaced transfusion therapy. The use of G-CSF and GM-CSF for mobilizing marrow progenitor cells and the effectiveness of G-CSF for granulocyte mobilization have been discussed in Chapter 14. Use of recombinant human erythropoietin (rhEPO) as an adjunct to autologous transfusions has been discussed previously in this chapter.
Erythropoietin
Erythropoietin (EPO) is a 36-kDa glycoprotein that is the primary regulator of erythropoiesis. EPO is produced primarily by the peritubular cells in the kidney in response to hypoxia. A small amount is made in the liver in adults. The EPO gene was cloned in 1985. Clinical-grade recombinant human erythropoietin (rhEPO) became available and became a standard treatment for anaemic patients with end-stage renal disease by 1989 (Fisher 2003). Administration of rhEPO results in egress of immature reticulocytes from the bone marrow and gradual elevation of the haematocrit (Spivak 1993, 2001). The drug is now administered in a variety of settings in addition to uraemia where it may replace the use of allogeneic blood (Cazzola et al. 1997). In the USA, the use of rhEPO almost immediately decreased consumption of red cells by 0.5 million units per year (Adamson 1991a,b). Darbopoietin alfa, a longer-acting erythropoietic agent, is a 165-amino acid protein that differs from recombinant human erythropoietin in containing 5 N-linked oligosaccharide chains, compared to the 3 chains of rhEPO. The additional carbohydrate chains increase the approximate molecular weight of the glycoprotein from 30 000 to 37 000 daltons. Darbopoietin is reportedly as effective as rhEPO at maintaining haemoglobin level but with less frequent dosing (Locatelli et al. 2001).
Treatment of Patients with Renal Failure: Curb Your Enthusiasm
The major cause of anaemia in patients with end-stage renal disease is a lack of production of erythropoietin. Replacing endogenous erythropoitin with an equivalent recombinant agent and using normalization of haematocrit as an endpoint seems intuitive.Recombinant human erythropoietin has been used for decades to treat the anaemia in these patients (Winearls et al. 1986; Eschbach et al. 1989). The standard dose of rhEPO in renal failure is 50–150 U/kg intravenously or subcutaneously three times a week until the haematocrit reaches 0.30–0.34 (Eschbach 2002). Darbepoetin alfa has been substituted with a starting dose of 0.45 mcg/kg weekly, either intravenously or subcutaneously. Dosage and administration interval (as long as 3 weeks) are ordinarily adjusted according to response.The clinical response to rhEPO will probably not improve at doses above 500 U/kg intravenously three times weekly. On average, subcutaneous (s.c.) administration is more effective than i.v., as the drug is released more slowly from the tissues, which results in a longer circulating half-life and lower but more sustained plasma levels (Sisk et al. 1991). Pharmacokinetic studies indicate that rhEPO has a half-life of 4–9 h after i.v. administration, but >24 h after s.c. injection. Treatment with rhEPO leads to functional iron deficiency as red cell production outstrips the mobilization of iron stores, and patients must be monitored appropriately (Brugnara et al. 1993). A randomized, open label study comparing intravenous iron with oral iron supplementation and controls in 157 cancer patients with chemotherapy-related anaemia suggested that intravenous iron raises haematocrit more effectively than does ferrous sulphate (Auerbach et al. 2004). Although patient randomization was not stratified for a variety of clinical factors, the study supports the notion that iron is necessary for at least a portion of patients treated with rhEPO.
The major side-effects of treatment with rhEPO are hypertension and thrombotic episodes, originally thought to be related primarily to excessive doses and too rapid correction of anaemia (Johnson et al. 1990; Spivak 2001). However, rhEPO may have other mechanisms of action and a haematocrit-independent, vasoconstrictive activity that can result in hypertension has been proposed (Bode-Boger et al. 1996; Banerjee et al. 2000). Although it seemed logical that correcting haematocrit to ‘normal’ would improve health and well-being in anaemic patients, early studies did not attempt to define an ‘optimum haematocrit’ value. As clinical studies have evolved, the target haematocrit for patients with chronic renal failure (CRF) has become controversial. The Normal Hematocrit Study provided an early indication that administration of rhEPO to raise Hb concentrations into the normal range could cause harm (Besarab et al. 1998). This randomized trial of 1233 CRF patients with cardiovascular disease was halted early when an interim analysis raised concerns about the risk of death and myocardial infarction in the normal Hct (42%) arm. The CHOIR trial, which randomized 1430 non-dialysis patients with CRF and no known cardiovascular disease likewise terminated early when a composite endpoint that included mortality, stroke, and myocardial infarction suggested increased risk in the normal Hb (13.5 g/dl) arm (Singh et al. 2006). It seems contradictory that in both trials, higher Hct is associated with fewer cardiovascular events. A meta-analysis of nine trials indicated that outcomes favoured lower, rather than higher, haemoglobin target concentrations (Phrommintikul et al. 2007). Finally, in the TREAT study a placebo-controlled trial of more than 4000 subjects with type 2 diabetes mellitus and CRF, darbopoietin alfa used to treat anemia and reduce the risk of death and major cardiovascular events in patients did not result in benefit; there was a significant increase in the incidence of stroke in the darbepoetin alfa group as compared with the placebo group and a significantly higher rate of thromboembolic events (Pfeffer et al. 2009). One hypothesis suggests that aggressive treatment with erythropoietic agents results in a rise in Hct that is too precipitous and too high in addition to oscillation in Hb concentration that might give rise to haemodynamic or rheologic instability (Unger et al. 2010). Given the current state of knowledge, erythropoietic stimulation should be administered cautiously in patients with CRF and it seems prudent to refrain from complete correction of anaemia.
EPO-related pure red cell aplasia has been reported in association with the appearance of circulating EPO-neutralizing antibodies (Casadevall et al. 2001; Bennett et al. 2004). Incidence is 10-fold higher when the Eprex® formulation is administered (Cournoyer et al. 2004). Discontinuation of drug and immunosuppressive therapy are associated with haematological recovery, and a majority (56%) regain responsiveness to rhEPO therapy, 89% of those without evidence of antibody at the time of re-exposure (Bennett et al. 2005).
Treatment of Patients without Renal Disease
Those patients with AIDS and zidovudine-induced anaemia, whose levels of erythropietin are decreased (<500 mu/ml), respond to treatment with rHuEPO (100 U/kg three times a week (Spivak et al. 1989). Good results have also been obtained in anaemia related to chemotherapy in patients with cancer whose EPO level is <200 mu/ml (Rizzo et al. 2010). The dose advised is 100–150 U/kg three times weekly (Miller et al. 1990). In a randomized, double-blind, placebo-controlled study of 375 patients with anaemia receiving nonplatin chemotherapy for solid or non-myeloid haematological malignancies, decreased transfusion requirements and increased haematocrit followed rhEPO treatment of 150–300 IU/kg three times per week subcutaneously for 12–24 weeks (Littlewood et al. 2001). rhEPO has been used for bone marrow transplantation (Steegmann et al. 1992), myelodysplastic syndrome (Di Raimondo et al. 1996), rheumatoid arthritis (Pincus et al. 1990) and the anaemia of prematurity (Bader et al. 1996; Maier et al. 2002).
A recent concern has been raised, and appropriately so, regarding the long-term repeated use of rhEPO, especially by patients with cancer. The expression of functional EPO receptors has been demonstrated in many human cancer cells where, at least in vitro, rhEPO can stimulate cell growth and survival and may induce resistance to selected therapies. Studies of patients with head and neck tumours and breast cancer suggested that rhEPO may play a role in decreased tumour control, overall survival, and risk of adverse vascular events (Henke et al. 2003; Leyland-Jones et al. 2005). A meta-analysis of 14 000 patients receiving rhEPO showed an increased mortality of 17% and a decreased survival of 6% (Bohlius et al. 2009). Current recommendations for patients undergoing myelosuppressive chemotherapy who have hemoglobin (Hb) less than 10 g/dl, emphasize balancing the potential risks such as thromboembolism and shorter survival with the benefit of avoiding blood transfusion (Rizzo et al. 2010). If used, it is prudent to select the lowest dose necessary toavoid transfusions. At the very least, clinical experience underlines the need for caution when this drug is considered for treating patients with cancer.
Colony-Stimulating Factors
Myeloid Growth Factors
Filgrastim is a human granulocyte colony-stimulating factor (G-CSF, Neupogen), produced by recombinant DNA technology. Filgrastim, the recombinant methionyl human granulocyte colony-stimulating factor (r-metHuG-CSF) is a 175 amino acid protein with a MW of 18 800 daltons and an amino acid sequence identical to the natural sequence except for the addition of an N-terminal methionine necessary for its expression in E coli. Filgrastim administration results in a dose-dependent increases in white cell counts, increased circulating segmented neutrophils, and an increased myeloid:erythroid ratio in the bone marrow (Gabrilove et al. 1988). G-CSF is not species specific and appears to have minimal effects on other hematopoietic cell types.
For use as mobilizing agents for granulocyte and progenitor cell collection, see Chapter 14.
Febrile Neutropenia with Cancer Chemotherapy
Febrile neutropenia with resultant infection remains among the most serious treatment-related toxicities of cancer chemotherapy, with a mortality rate ranging from 2–21% (Smith et al. 2006). The risk of febrile neutropenia and subsequent infection is directly related to the duration and severity of neutropenia (Bodey et al. 1966; Bodey 1986). Filgrastim has been shown to accelerate the recovery of neutrophil counts following a variety of chemotherapy regimens.
More than a dozen studies have evaluated the benefit of administering myeloid cytokines (granulocyte colony-stimulating factor, G-CSF; granulocyte–macrophage colony-stimulating factor, GM-CSF) to febrile neutropenic patients with myeloid and non-myeloid malignancies. Although such therapy is more convenient than granulocyte transfusion, the results are scarcely more satisfying. A Cochrane analysis found that overall mortality and disease progression were not influenced by the use of colony-stimulating factor (CSF) (Clark et al. 2003). The benefit in reducing infection-related mortality is marginal, and even this result was highly influenced by one study. The prophylactic use of these cytokines for patients with febrile neutropenia due to cancer chemotherapy can reduce IV antibiotic use, the period of severe neutropenia, and the number of hospital days. Although primary prophylaxis with a myeloid growth factor can reduce the incidence of febrile neutropenia by as much as 50%, such use is hard to justify if there is no improvement in response or survival (Ozer et al. 2000). The collective results of eight treatment trials provide consistent support for the recommendation that cytokines should not be used routinely as adjunctive therapy for the treatment of uncomplicated fever and neutropenia (Ozer et al. 2000). Nevertheless, current ASCO guidelines recommend administration of myeloid cytokines in clinical settings where the expected risk of suffering febrile neutropenia is approximately 20%. Secondary prophylaxis with myeloid cytokines is recommended for patients who have developed a neutropenic complication during a previous chemotherapy cycle and in whom a reduced chemotherapy dose might compromise disease-free or overall survival (Smith et al. 2006). The experience is similar for patients undergoing stem cell transplants For patients with myeloid and non-myeloid malignancies treated with myeloablative therapy and allogeneic stem cell transplants, a significant reduction in the median number of days of severe neutropenia can be achieved, but there is no evidence of a survival advantage for these patients.
Chronic Neutropenia
Severe chronic neutropenia, an absolute neutrophil count of less than 0.5 × 109/l lasting from months to years, was one of the original indications for G-CSF treatment. Congenital, cyclic and idiopathic neutropenia fall into this category. A sustained increase of the neutrophil count, a reduction of the number of infections and reduced requirement for antibiotics was obtained in 40 out of 44 children with congenital neutropenia. Treatment for 4–6 years was well tolerated in the majority of patients and resulted in a long-term improvement of the clinical condition (Bonilla et al. 1989). More than 850 patients, most treated with daily or alternate-day recombinant human G-CSF (or filgrastim), have been followed on the Chronic Neutropenia Registry (Dale et al. 2003). G-CSF treatment increased the ANC 10-fold during the first year of treatment. For most patients, the responses were durable and patients remained on the same dose of G-CSF for years. Most patients remained free of bacterial infection. Thrombocytopenia developed in 4% of patients and myelodysplasia or acute myelocytic leukaemia has occurred in 35 out of 387 patients with congenital neutropenia with a cumulative risk of 13% after 8 years of G-CSF treatment. These events occurred without a predictable relationship to the duration or dose of G-CSF treatment. It is not clear whether the drug had any causative role in these disorders. No patient with cyclic or idiopathic neutropenia developed myelodysplasia or leukaemia. Growth and development and the outcome of pregnancy appeared to be unaffected by G-CSF treatment (Dale et al. 2003).
Mild-to-moderate bone pain was reported in approximately 33% of patients on chronic G-CSF treatment. Pain is readily controlled with non-narcotic analgesics. Palpable splenomegaly was observed in approximately 30% of patients. Abdominal or flank pain was seen infrequently, and thrombocytopenia was noted in 12% of patients with palpable spleens. Fewer than 3% of all patients underwent splenectomy, and most of these had a prestudy history of splenomegaly.
Neonatal Neutropenia
Sepsis is a major cause of neonatal death. Published rates of sepsis in preterm newborns range from 25–50% and neonates in whom late onset sepsis develops are significantly more likely to die than those who avoid infection (Stoll et al. 1996; Cooke et al. 1997). In infants of very low birth-weight, GM-CSF has been found to produce a significant increase in the neutrophil and platelet counts and in the bone marrow neutrophil storage pool (Cairo et al. 1995). However in a randomized trial for preventing sepsis in small-for-gestational age preterm neonates, GM-CSF did not reduce sepsis-free survival at 14 days, even though it increased the neutrophil count (Carr et al. 2009). Whereas the trial can be criticized for being underpowered, for using the wrong cytokine, or even the wrong endpoint, it once again emphasizes the folly of relying on surrogate markers, even such physiologically rational ones as neutrophil elevation, instead of clinical response.
Thrombopoietin
For more than 50 years scientists searched for ‘thrombopoietin’, the humoral factor that regulates platelet production and a potential treatment for patients with severe thrombocytopenia (Kelemen et al. 1958). Non-lineage specific cytokines such as interleukin 6 (IL-6) and interleukin 11( IL-11) can stimulate platelet production; IL-11 has been used to reduce the need for platelet transfusions in patients with chemotherapy-induced thrombocytopenia (Gordon et al. 1996; Vredenburgh et al. 1998). These recombinant proteins are not true thrombopoietins. Although IL-11 administration can reduce the need for platelet transfusions in selected patients with severe thrombocytopenia, treatment is accompanied by disturbing side-effects such as fever, fatigue, chills, dyspnoea, hypotension, peripheral oedema, atrial arrhythmias and syncope.
In 1994 human thrombopoietin (TPO), also called c-Mpl ligand, was purified and cloned. TPO proved to be a 95-kDa, 332-amino-acid, lineage-specific glycoprotein which stimulates megakaryocyte growth and maturation and has considerable homology to erythropoietin (de Sauvage et al. 1994; Foster et al. 1994; Lok et al. 1994). The TPO receptor, c-MPL, contains a dimeric structure which likely changes shape with TPO binding and activates the JAK2-STAT5 signal transduction pathway (Remy et al. 1999). TPO binds to a variety of hematopoietic cells, but it does not bind to the EPO receptor. TPO is synthesized primarily in the liver (Peck-Radosavljevic et al. 2000). Various recombinant preparations of TPO have been developed including recombinant human thrombopoietin (rhTPO) and pegylated recombinant human megakaryocyte growth and development factor (PEG-rHuMGDF). Plasma levels of TPO usually increase as platelet mass declines, and remain elevated during the course of thrombocytopenia. Platelet transfusions ordinarily decrease the plasma TPO levels, as TPO binds to the c-Mpl receptor on platelets and is removed from the circulation (Scheding et al. 2002).
Studies in Normal Volunteers: Some Ups and Unexpected Downs
Two studies with these ‘first-generation’ thrombopoietins were conducted with healthy human volunteers. In the first, thrombapheresis donors were treated with a single dose of PEG-rHuMGDF and underwent apheresis 14 days later (Kuter et al. 2001). There was a dose-dependent rise in platelet count and a proportionate increase in aphaeresis yield (Figure 18.1).
Figure 18.1 PEG-rHuMGDF increases the yield of platelet apheresis. Administration of a single dose of PEG-rHuMGDF produced a dose-dependent increase in platelet count and apheresis yield 15 days later. (a) The platelet count rises in a dose-dependent manner following the administration of PEG-rHuMGDF to apheresis donors. The response of the donor median platelet count (+ SD) to placebo ( ), 1 µg/kg PEG-rHuMGDF (
), 1 µg/kg PEG-rHuMGDF ( ), or 3 µg/kg PEG-rHuMGDF (
), or 3 µg/kg PEG-rHuMGDF (  ) administration. Platelet counts in donors were obtained on day 1 just prior to injection, on the indicated days thereafter, before (15, pre) and after (15, post) platelet apheresis. (b) Platelet apheresis yield is directly proportional to the donor preapheresis platelet count. The relationship between the preapheresis platelet count on day 15 and the platelet apheresis yield is shown for donors treated with placebo (
) administration. Platelet counts in donors were obtained on day 1 just prior to injection, on the indicated days thereafter, before (15, pre) and after (15, post) platelet apheresis. (b) Platelet apheresis yield is directly proportional to the donor preapheresis platelet count. The relationship between the preapheresis platelet count on day 15 and the platelet apheresis yield is shown for donors treated with placebo ( , n = 65), 1 µg/kg PEG-rHuMGDF (
, n = 65), 1 µg/kg PEG-rHuMGDF ( , n = 23), or 3 µg/kg PEG-rHuMGDF (•, n = 22).
, n = 23), or 3 µg/kg PEG-rHuMGDF (•, n = 22).
(Source: Kuter et al. 2001. Reproduced with permission of the American Society of Hematology.)

No adverse events were noted. The aphaeresis platelets reacted normally in aggregation studies and resulted in greater platelet count increments and longer transfusion-free intervals than did platelets obtained from donors treated with placebo (Goodnough et al. 2001) TPO-generated autologous platelets have also been cryopreserved and later transfused into their donors after chemotherapy (Vadhan-Raj et al. 2000). In the second study, 538 paid healthy subjects received up to 3 doses of 3 µg/kg PEG-rHuMGDF every 28 days (Li et al. 2001).The peak platelet count rose to 589 × 109/L on the first cycle, compared with the baseline counts of 240 × 109/L, but declined with each subsequent cycle (506 × 109/l and 494 × 109/l).Unfortunately, 13 subjects treated with PEG-rHuMGDF (0 of 210 after 1 dose, 2 of 204 after 2 doses, and 11 of 124 after 3 doses) developed thrombocytopenia and produced an antibody against PEG-rHuMGDF that cross-reacted with endogenous thrombopoietin (see below).
Chemotherapy-Induced Thrombocytopenia
Of the two recombinant thrombopoietins, PEG-rHuMGDF, the most widely studied, produces dose-dependent increases in platelet counts in patients with advanced malignancies and chemotherapy-induced thrombocytopenia (Basser et al. 1997; Fanucchi et al. 1997). When administered before chemotherapy as a daily subcutaneous injection, PEG-rHuMGDF produced a dose dependent increase in peripheral blood platelet count. No evidence of platelet activation or altered platelet function has been observed. In a randomized, dose-escalation study, the platelet nadir in 53 patients with lung cancer treated with PEG-rHuMGDF after chemotherapy was higher than that of control subjects, but the need for platelet transfusions was unchanged because few patients developed severe thrombocytopenia (Basser et al. 1997). Similar results have been reported in other studies of chemotherapy for non-myeloid malignancies. Some reduction in the need for platelet transfusion may be found, but the effect is usually limited to the early cycles of chemotherapy.
The experience has been comparable with recombinant human thrombopoietin. When administered as a single intravenous dose before chemotherapy, rhTPO was associated with a dose-dependent increase in platelets that began about day 4 and peaked about day 12 (Vadhan-Raj et al. 1997). rhTPO administered subcutaneously to previously treated patients with gynaecological malignancies before and after chemotherapy produced a modest dose-dependent rise in circulating platelet count. The need for platelet transfusions decreased by 75% (Vadhan-Raj et al. 1997). PEG-rHuMGDF and rhTPO have not improved thrombocytopenia significantly when administered to patients receiving intensive chemotherapy for acute leukaemia or stem cell transplantation, and failed to reduce the requirement for platelet transfusion (Nash et al. 2000; Schiffer et al. 2000). However timing and dose are critical factors, and neither has been studied extensively in this setting.
Stem Cell Transplants and Thrombocytopenia
Administration of either PEG-rHuMGDF or rhTPO to patients who received an autologous stem cell transplant had no effect lowest platelet count, the platelet recovery interval, or number of platelet transfusions (Nash et al. 2000; Schuster et al. 2002). It is unclear whether this lack of response was due to the lack of target bone marrow cells, the existing high levels of TPO, or other unidentified factors. Nonetheless, in repeated small studies in which both the dose and schedule of the thrombopoietic growth factors were altered, there appeared to be no benefit. The only clinical benefit shown with thrombopoietic growth factors in stem cell transplantation occurred when the stem cell donor was treated with rhTPO.
Immune Thrombocytopenia
Immune thrombocytopenic purpura (ITP), a disorder of increased peripheral platelet destruction, is discovered increasingly as an incidental finding of mild to moderate thrombocytopenia. ITP is currently defined as an immune disorderin which an isolated platelet count of <100 × 109/l is found without another implicated disorder. Patients still present with petechiae, easy bruising, or mucous membrane bleeding as a consequence of severe thrombocytopenia. Severely thrombocytopenic patients (platelets <1000/µl) are at increased risk of intracranial haemorrhage, particularly if they sustain head trauma (George et al. 1996). Transfused platelets usually result in small increments and short survival. (Abrahm and Ellman 1976; Carr et al. 1986). Platelet transfusions play a limited therapeutic role in this disorder (see Chapter 14). Whereas the classic teaching emphasized destruction and clearance of antibody-coated platelets, the pathogenesis of ITP is clearly much more complicated. Impaired or insufficient platelet production is now well recognized as contributing to the pathogenesis of ITP in many patients (Ballem et al. 1987). More recent investigations implicate disordered cellular immunity as well.
Decreased production is generally attributed to a direct effect of antibody on megakaryocyte maturation or platelet release. By stimulating platelet production and release, TPO administration increases circulating platelets in some ITP patients. Six HIV-infected patients with ITP and normal or slightly elevated endogenous TPO levels experienced a 10-fold rise in platelet count within 14 days of the start of PEGrHuMGDF (Cole et al. 1998). Similar responses have been reported in patients with non-HIV-related ITP treated with intravenous and subcutaneous TPO, in some instances for years. These patients likely have a suboptimal endogenous TPO response, possibly as a result of their reduced megakaryocyte mass, but increase platelet production when recombinant drug is administered.
Safety of Recombinant Thrombopoietin
Administration of multiple doses of first-generation recombinant thrombopoietins to some cancer patients and healthy volunteers has been associated with the development of cross-reacting antibodies and thrombocytopenia (Li et al. 2001). Spontaneous TPO autoantibodies with thrombocytopenia have also been reported (Shiozaki et al. 2000). Thrombocytopenia occurred in 4 out of 665 oncology patients administered multiple doses, in 2 of 210 healthy volunteers who received two doses and in 11 out of 124 healthy volunteers given three doses of PEG-rHuMGDF (Li et al. 2001). No subject developed antibodies or thrombocytopenia after a single injection. These observations ended further clinical studies of the first-generation thrombopoietic growth factors. Nevertheless, the lessons learned in these studies are valuable for evaluating other TPO-receptor agonists that act through the JAK2-STAT5 pathway.
Second Generation Thrombopoietins
The encouraging clinical results with first generation thrombopoietins motivated a search for agents that stimulate platelet production without eliciting antibodies that crossreact with endogenous TPO. The most promising candidates include TPO peptide mimetics, TPO non-peptide mimetics, and TPO-agonist antibodies (Kuter 2010). All of these bind to and activate the TPO receptor and all have unique pharmacologic attributes.
AMG 531(Romiplostim, Nplate)
AMG 531 is a rationally designed TPO peptide-agonist found by screening peptide libraries to bind to the TPO receptor and composed of an Fc antibody fragment bound to two identical peptide sequences linked via polyglycine (‘peptibody’). The peptide residues form four TPO receptor binding sites, but they display no amino acid sequence homology to TPO. The Fc component extends the half-life of the molecule to 110–160 hours; after binding and internalization, it is removed by the reticuloendothelial system (Kuter 2010). There is little experience in patients with hepatic or renal impairment. Because Romiplostim has no sequence homology with TPO, antibodies formed against this agent should not cross react with endogenous TPO.
A single intravenous or subcutaneous administration of Romiplostim induces a dose-dependent increase in platelet count in healthy subjects, with peak platelet count between days 12 and 16 (Wang et al. 2004). The highest intravenous dose, 10.0 µg/kg, caused a nearly 6-fold increase in platelet count, >1000 × 109/l. Of eight subjects receiving a 2.0-µg/kg subcutaneous dose, six had peak platelet levels that were double the baseline value. Platelet response was similar whether after single intravenous or subcutaneous administrations of Romiplostim at the same dose level (1.0 microg/kg), even though subcutaneous serum levels were barely detectable, and in either case returned to baseline by day 28. The most frequent side effects were mild to moderate headache and sore throat. No serious adverse events were reported in this study, nor were antibodies to TPO detected. In its current formulation, Romiplostim is administered as a weekly subcutaneous injection.
In an early study of Romiplostim for ITP, 7 of 12 patients doubled their platelet count with increases above 50 × 109/l after 2 doses of 3, 6, or 10 µg/kg. The peak platelet count was dose dependent: 163 × 109/l, 309 × 109/l, and 746 × 109/l for the 3, 6, and 10 µg/kg cohorts, respectively. In a later study, Romiplostim (1 or 3 µg/kg) or placebo were injected weekly for 6 weeks. Twelve of 16 patients treated had their platelet counts double and rise to greater than 50 × 109/l. Mean peak platelet counts were 135 × 109/l, 241 × 109/l, and 81 × 109/l for the 1 µg/kg, 3 µg/kg, and placebo groups, respectively (Bussel et al. 2006). Analysis of 36 patients, most treated for more than 48 weeks with weekly injections of Romiplostim, showed a stable mean platelet count of greater than 100 × 109/l, and half were able to stop other concomitant immunosuppressive drugs (Kuter et al. 2008). On stopping therapy, all of the subjects had their platelet count return to baseline. A few had a rebound worsening of their baseline thrombocytopenia lasting for up to 2 weeks. The only potential long-term complication that was identified was increased bone marrow reticulin noted in 2 of more than 100 patients.
Repeated weekly dosing of Romiplostim over a 24-week period in two placebo-controlled studies of 63 splenectomized and 62 non-splenectomized chronic patients ITP led to an overall response (platelet counts ≥50 × 109/l during ≥4 weeks on study) in 83% of patients and a durable response in 49% of patients (Kuter et al. 2008). A durable platelet response was achieved by 16 of 42 splenectomized patients given Romplostim, but none of 21 given placebo, and by 25 of 41 non-splenectomized patients given Romplostim versus one of 21 given placebo.
In an extension study of 142 subjects who participated in previous Romiplostim trials, patients were treated for a mean of 69 weeks (maximum 3 years). Platelet response (count greater than or equal to 50 × 109/l and a doubling from baseline) was achieved in 87% of subjects and sustained for the duration of study (Bussel et al. 2009). Concerns persist about the safety of long-term Romiplostim use, particularly with respect to the risk of bone marrow fibrosis.
Eltrombopag (Promacta)
Eltrombopag belongs to the bioarylhydrazone class of non-peptide small molecules and is an oral TPO-receptor agonist. Eltrombopag acts at a site distant from the TPO binding site, apparently involving histidine 499 in the transmembrane region, and does not compete with endogenous TPO. It stimulates megakaryocytes to proliferate and differentiate and results in a dose-dependent increase in circulating platelets. Unlike Romiplostim, a single dose of Eltrombopag results in no increase in platelet count, however when administered as daily doses of 50–75 mg, the platelet count begins to rise on days 8–10 and peaks about day 16, similar to what is seen with Romiplostim (Jenkins et al. 2007). Metabolism and elimination is about 2/3 hepatic and 1/3 renal. Eltrombopag has low immunogenic potential.
Eltrombopag is indicated for patients with chronic ITP and increased risk of bleeding, particularly after conventional therapies have failed. At a dose of 75 mg, some 80% of patients increase their platelet count to >50 000/µL and another 20% to 30% will increase their platelet count to >200 000/µL over a period of 2–3 weeks. A randomized study of chronic ITP (platelet count 30 000/µL) after failed previous treatment, patients showed a significant benefit for eltrombopag compared to placebo, when elevation of platelet count to >50 000 after 6 weeks was used as an endpoint, and a significant reduction in the proportion of patients who bled (Bussel et al. 2009). Platelet count returned to baseline about 2 weeks after cessation of treatment. Remarkably, response rate did not appear to be influenced by concomitant ITP medications, absolute platelet count, splenectomy status, or the number of previous ITP therapies. The major side effects were nausea and vomiting.
Both synthetic TPO-receptor agonists are indicated as treatment for chronic ITP. Comparing Romiplostim to eltrombopag, one drug is injectable and the other oral. However the injection is administered once a week, whereas the oral drug is taken daily. Although both drugs are used primarily for ITP, there is evidence of benefit for patients with aplastic anaemia, myelodysplasia, and thrombocytopenia related to chronic liver disease and chemotherapy (Vadhan-Raj 2010; McHutchison et al. 2007).
Adjuncts to Haemostasis
Pharmaceutical and biological agents occasionally replace transfusion therapy, but are more commonly used as adjuncts in the treatment of patients with haemostatic disorders (Mannucci 1998). A wide range of agents is available. Recombinant factor VIIa has emerged not as a replacement therapy, although it is licensed for this purpose in the US and Europe, but as a treatment for bleeding haemophiliacs with inhibitors (Chapter 14), and as both prophylaxis and therapy for patients with massive bleeding. A synthetic analogue of L-vasopressin, DDAVP, has long been used to minimize haemorrhage for most mild forms of haemophilia A and VWD, and is increasingly recognized to have multiple less well-defined haemostatic effects when administered empirically in other circumstances. Lysine analogues that inhibit fibrinolysis are used both systemically and locally for acquired and inherited defects in haemostasis. Vitamin preparations in the naphthoquinone family (vitamin K) are used to prevent neonatal bleeding syndromes as well as to reverse warfarin anticoagulation or to treat ingestion of warfarin-like rodenticides. Still other agents such as oestrogens and protamine sulphate have proved valuable adjuncts to transfusion therapy. On the other hand, Aprotinin, a bovine-derived serine protease inhibitor with potent antifibrinolytic activity, was widely employed to enhance surgical haemostasis after cardiopulmonary bypass (as described in the previous edition), but has now been removed from the market because of newly-appreciated toxicities. Perhaps the lesson here is that blood components with all of the recognized and potential risks have been studied for almost a century and administered to hundreds of millions of recipients; pharmacologic replacements and adjuncts, however promising, may well eventually display toxicities that cannot be recognized in the limited number of subjects treated in clinical trials and even after several year’s worth of clinical experience.
Recombinant Factor VIIa
The effectiveness of plasma fractions containing activated clotting factors in treating factor VII-deficient patients as well as other haemophiliacs with coagulation factor inhibitors led to the appreciation of the singular role of factor VII and the development of recombinant factor VIIa (rFVIIa) for human use (see also Chapter 14) (Hedner and Kisiel 1983). Recombinant factor VIIa, a two-chain procoagulant enzyme of approximately 50 000 molecular weight, becomes active when complexed with tissue factor in the extrinsic clotting cascade (Butenas et al. 2003). The circulating half-life is between 2 and 3 h, although it appears to be somewhat shorter in children than in adults (Villar et al. 2004). Dosage remains contentious. Most studies have used a dose of 90 µg/kg repeated every 2 h; however, doses as low as 30 µg/kg and as high as 120 µg/kg have been administered. The mechanism of action remains controversial. Preclinical studies in dogs indicate that functional platelets are critical to achieve normal haemostasis, although FVIIa can induce localized fibrin deposition on the surface of platelets with defective aggregation and induce platelet aggregates (Lisman et al. 2004). At pharmacological doses, rFVIIa binds directly to activated platelets and may generate a burst of thrombin that is localized to the site of bleeding (Lisman et al. 2004). For this reason, administration of rFVIIa has generally not been recommended for thrombocytopenic patients. However rFVIIa also complexes directly with tissue factor (TF) released from the subendothelium at sites of vascular injury. The TF–rFVIIa complex can activate the common coagulation cascade via activated factor X (Hedner 2008). Anecdotal reports suggest that even with severe, refractory thrombocytopenia, haemostasis may be improved (Vidarsson and Onundarson 2000; Gerotziafas et al. 2002; Tranholm et al. 2003). Whether the absolute platelet count or the mechanism of thrombocytopenia is important has yet to be determined.
Additional Indications
Recombinant VIIa has been administered for a wide range of bleeding problems in addition to replacement therapy and management of haemophiliacs with inhibitors. The drug reduces the prothrombin time and controls bleeding in patients who require rapid reversal of warfarin anticoagulation (Deveras and Kessler 2002; Freeman et al. 2004). Changes in the laboratory test do not correlate with cessation of bleeding. There is no assay suitable for monitoring drug efficacy. rFVIIa infusion may provide one option for reversing treatment with newer anticoagulants such as those that target thrombin, factor Xa, or tissue factor, but as yet little experience has been reported in this setting (Levi et al. 2004). rVIIa has reportedly controlled bleeding in a variety of inherited and acquired disorders of platelet function, such as Glanzmann thrombasthenia, Hermansky–Pudlak syndrome, Bernard–Soulier syndrome, VWD and uraemia (Monroe et al. 2000; Pozo et al. 2002; Poon et al. 2004). Patients with liver disease and profuse variceal bleeding have responded promptly to bolus infusions and the drug has been used prophylactically for patients undergoing liver biopsy (Bosch et al. 2004; Bosch et al. 2008; Caldwell et al. 2004; Romero-Castro et al. 2004). Although bleeding has been well controlled in a subgroup of patients with variceal haemorrhage, reduction in mortality has not yet been seen. rFVIIa is reportedly effective and well tolerated as an option for managing central nervous system bleeding in patients with VII deficiency, anticoagulation, haemophilia with inhibitors and possibly other conditions such as aneurysms and trauma (Schmidt et al. 1994; Freeman et al. 2004; Huang et al. 2004). In a proof-of-concept study, rVIIa (160 µg/kg) administered within 4 h of symptoms reduced haematoma expansion at 24 h in patients who sustained intracerebral haemorrhage and improved survival at 90 days.(Mayer et al. 2005) Each of three treatment groups realized improvements in functional scores when compared to a placebo control group at 3 months. However a larger study by the same investigators failed to find a mortality or morbidity advantage in the treated group, despite the evident reduction in intracerebral haematoma expansion.(Mayer et al. 2008). Arterial thromboembolic events were more common in the treatment groups.
Trauma and Surgery
The earliest and perhaps most promising applications for rVIIa seemed to be in the settings of trauma and surgery with profuse bleeding. The observation that after a bolus of rVIIa, bleeding may cease virtually instantaneously in patients with desperate wounds or profuse bleeding at operation has produced many advocates but few controlled trials (Kenet et al. 1999; Martinowitz et al. 2001). In a series of trauma patients, coagulopathy was reversed in 61 out of 81 cases, with an associated reduction in prothrombin time from 19.6 to 10.8 s. However, these observations did not translate into improved mortality compared to historical controls (Dutton et al. 2004). Two multi-centre randomized trials, one in the setting of blunt trauma (69 patients), the other with penetrating trauma (70 patients), showed some reduction in red cell transfusion and in the frequency of massive transfusion, but no reduction in the rates of multiorgan system failure, the duration of intensive care unit stay, or mortality (Boffard et al. 2006). A later trial of similar design involving 150 international sites to study the role of rFVIIa in blunt and penetrating trauma was terminated early on the basis of a statistical prediction of futility. The only RCT addressing the efficacy of rFVIIa in the management of traumatic brain injuries compared multiple dose regimens with placebo for 61 patients with trauma-induced intracerebral hemorrhage at 38 centres. Although patients receiving more than 80 µg/kg of rFVIIa trended toward decreased hematoma volume, no significant differences in Glasgow Coma Scores or mortality rates were found (Narayan et al. 2008).
Similar dramatic improvements in haemostasis have been reported anecdotally with cardiac surgery and surgery for extensive, disfiguring facial haemangiomas (Tanaka et al. 2003; Waner 2004). Randomized trials have not supported these findings. A small trial in which 20 patients were randomized to placebo or treatment with a single dose of rFVIIa (90 µg/kg) after the reversal of heparinization with protamine found that the number of patients requiring red cell transfusion was significantly lower in the treatment group, but no other statistically significant differences between the groups (Diprose et al. 2005). A larger, multidose rVIIa trial reported similar findings as well as a caution regarding potential drug toxicity (Gill et al. 2009). In the sole pediatric trial, a study of prophylactic rFVIIa during and after surgical correction of congenital heart defects in 76 infants younger than age one, rFVIIa administration appeared (paradoxically) to increase the time from heparin reversal to chest closure, the study primary endpoint, and failed to reduce transfusion of red cells, platelets, or plasma (Ekert et al. 2006). In a controlled trial of rVIIa during retropubic prostatectomy, the patients who received drug sustained less blood loss and required on the average fewer red cell transfusions (Friederich et al. 2003). In summary, trials in these settings indicate that rVIIa can reduce red cell transfusion, but not transfusion of other blood components, does not improve morbidity, mortality, or hospital stay, and does not reduce the cost of medical care.
One surprise has been the apparent dearth of side-effects related to the administration of an activated clotting factor. In about 800 000 administrations to patients with haemophilia over a three-year period, only a 1% incidence of serious adverse reactions has been reported (Abshire 2008). However studies of ‘off-label’ uses paint a more sobering picture. Data from 35 randomized clinical trials for different off-label uses of rVIIa (26 studies involving 4119 patients and 9 studies involving 349 healthy volunteers) reported 498 subjects with thrombotic events (Levi et al. 2010). Arterial thromboembolic events, particularly among subjects 65 years of age or older, were significantly more common among those who received rFVIIa than among those who received placebo, whereas rates of venous thromboembolic events were similar. This finding serves as a reminder that prohaemostatic agents have potential risk and must be administered judiciously, even in the face of life-threatening haemorrhage.
DDAVP (1-Deamino-8-D-Arginine Vasopressin or Desmopressin)
DDAVP is a synthetic analogue of the antidiuretic hormone L-vasopressin that has been used to control bleeding in patients with mild congenital or acquired bleeding disorders for more than 25 years (Richardson and Robinson 1985; Mannucci 1986; Lee et al. 1993). The drug has been used to reduce blood loss in a variety of surgical settings; however, with the possible exceptions of patients who have ingested aspirin or have other disorders of platelet function, benefit appears to be minimal (Dilthey et al. 1993; Despotis et al. 1999b; Carless et al. 2004).
Mechanisms of Action and Tachyphylaxis
DDAVP administration raises circulating FVIII and VWF levels, which account in part for the drug’s haemostatic effect (Edelson et al. 1974; Richardson and Robinson 1985). Compared with vasopressin, DDAVP has increased affinity for V-1 receptors, which results in rapid release of FVIII and VWF from preformed cellular stores, and markedly decreased affinity for V-2 receptors that mediate vasoconstriction (Richardson and Robinson 1985; Mannucci 1986). In normal subjects, DDAVP increases FVIII and VWF levels within 30 min after infusion. Levels peak at 300–400% of baseline in 1–2 h, and persist for 6–12 h (Mannucci et al. 1981). Response is faster with intravenous infusion than with intranasal administration.
Both red blood cell (RBC) and platelet adhesion to endothelial cells are increased by DDAVP when studied in vitro (Tsai et al. 1990; Rosse and Nishimura 2003). These effects might be produced by direct action on the vessel wall (Rosse and Nishimura 2003) or by release of high-molecular-weight VWF at the endothelial cell surface (Barnhart et al. 1983; Takeuchi et al. 1988). DDAVP also increases procoagulant platelet microparticle formation (Horstman et al. 1995), expression of tissue factor on endothelial cells (Galvez et al. 1997) and expression of p-selectin and the adhesive glycoprotein Ib on platelet membranes (Sloand et al. 1994; Wun et al. 1995). One or more of these or other unidentified effects may explain the observation that DDAVP shortens the bleeding time in patients with severe VWD who have already received cryoprecipitate infusions (Cattaneo et al. 1989), and in qualitative platelet disorders such as uraemia (Mannucci et al. 1983), liver disease (Mannucci et al. 1986) or other acquired or congenital conditions (Kentro et al. 1987) in which levels of FVIII and VWF are usually normal.
DDAVP infusion for haemophilia A and VWD produces short-term increases of circulating FVIII and VWF as preformed cell stores are released (Mannucci 1997). DDAVP alone has been used to control bleeding associated with minor surgical procedures or dental work, but is inadequate for procedures that require prolonged haemostasis. Repeat administration of DDAVP at intervals shorter than 24 h or repeatedly for several days is associated with decreased laboratory and clinical response (tachyphylaxis) (Mannucci et al. 1992; Mannucci 2004). The blunted response apparently results from depletion of intracytoplasmic stores of FVIII and VWF. The pattern of tachyphylaxis is not predictable. Patients with VWD are less likely to respond poorly or fail to respond after repeated doses. Although the haemostatic response to initial administration of DDAVP varies among patients, it is usually reproducible on each occasion for a given patient (Mannucci et al. 1992). If management with DDAVP is planned, patients should receive a test infusion to establish response several days prior to any planned invasive procedure.
Dose and Administration
DDAVP may be administered by intravenous, subcutaneous and intranasal routes. The intravenous dose is 0.3 µg/kg, administered over 30 min in 50 ml of normal saline for adults (in 10 ml for children weighing less than 10 kg). The maximum response, factor elevations of three to five times baseline levels, occur within 30 to 60 min at intravenous doses of 0.3 µg/kg (Mannucci et al. 1981, 1992). The subcutaneous dose is 0.3–0.4 µg/kg, with peak responses approximately 230% above baseline at 60 min after administration, slightly lower and later than the peak dose after intravenous administration. The intranasal dose is an order of magnitude higher than the intravenous or subcutaneous dose, 300 µg for adults. Higher doses do not enhance efficacy, but may be associated with increased toxicity. A concentrated nasal spray formulation is available in both the USA and Europe. However, a concentrated preparation for subcutaneous use is not available in the USA, and the substantial volume (7 ml) required for subcutaneous treatment of a 70-kg individual may be uncomfortable. DDAVP is cleared by the liver and kidneys and has a plasma half-life of 124 min (Mannucci 1997).
Indications
As the bleeding tendency in haemophilia A, and to a lesser degree in VWD, correlates with measured blood levels of the deficient factor, the ability of DDAVP to transiently raise FVIII and VWF has resulted in its approval for use in these disorders. Experience has confirmed its usefulness and DDAVP is now the treatment of choice for minor and even moderately invasive procedures in patients with these disorders who respond to a test infusion (Mannucci 1997; Porte and Leebeek 2002; Mannucci 2004). Response is estimated from prospective studies at about 28% for VWD type I, 18% for type IIA, 14% for type IIM and 75% for type IIN (Federici et al. 2004). Genotype predicts response better than does phenotype for type IIA and IIN. The use of DDAVP in type IIB VWD remains controversial, but is generally contraindicated because these patients develop mild thrombocytopenia related to the affinity of the VWF for the platelets. Infusion of DDAVP and release of endogenous VWF stores into circulation may worsen thrombocytopenia (Mannucci 2004). Nevertheless improved haemostasis in type IIB VWD after infusion of DDAVP associated with little or only mild thrombocytopenia has been reported (McKeown et al. 1996). In patients with severe haemophilia A, FVIII levels do not increase after DDAVP infusion; however, DDAVP may still provide potential benefit by increasing VWF levels, with an associated increased response in activity of infused FVIII concentrates (Deitcher et al. 1999).
DDAVP has been found to augment haemostasis in a variety of acquired and congenital conditions with impaired haemostasis in which other treatment options are limited. In a double-blind placebo-controlled study of patients with congenital platelet defects, DDAVP lowered the bleeding time most effectively in subjects with normal platelet-dense granule stores (Rao et al. 1995). In other studies, DDAVP has been shown to improve the bleeding time or be associated with adequate surgical haemostasis in patients with storage pool defect (Schulman et al. 1987; Kobrinsky et al. 1991), Bernard Soulier disease (Noris et al. 1998), aspirin ingestion (Flordal and Sahlin 1993; Reiter et al. 2003) and other defects in platelet haemostasis. DDAVP shortens the bleeding time in some haemorrhagic diseases of multifactorial origin such as cirrhosis and uraemia. Extrapolation of the utility of DDAVP in these conditions should be done with caution. Most studies have determined the efficacy of DDAVP by observing a shortening of the bleeding time, a notoriously poor predictor of haemostasis in most settings, or by assessing surgical haemostasis in uncontrolled studies of small numbers of patients.
Initial reports that DDAVP decreased blood loss and transfusion requirements after cardiac and spinal surgery have not been confirmed in follow-up studies (Mannucci 1997; Porte and Leebeek 2002). One large meta-analysis revealed no reduction in bleeding after the use of DDAVP in cardiac surgery (Brown et al. 2007). DDAVP had no effect on mortality or need for repeat thoracotomy, but was associated with a two- to four-fold increased risk of coronary thrombosis. A Cochrane analysis arrived at a similar conclusion (Carless et al. 2004). The subset of patients with preoperative platelet defects, including those that have ingested aspirin, appears to benefit from administration of DDAVP prior to surgery. Blood loss was markedly reduced in patients with preoperative defects in platelet function (identified by point-of-care testing) who received DDAVP compared with those who received placebo (Despotis et al. 1999b). Three randomized double-blind placebo-controlled trials have shown clinically significant reductions in blood loss and transfusion requirements after the use of DDAVP in patients who have ingested aspirin before cardiac surgery (Gratz et al. 1992; Dilthey et al. 1993; Sheridan et al. 1994). DDAVP also reduced bleeding in an unblinded comparison with placebo in patients who ingested aspirin before cholecystectomy (Gratz et al. 1992). In addition, DDAVP has been demonstrated to shorten the bleeding time in normal volunteers after aspirin ingestion, perhaps due to direct effects on platelets or increases in VWF (Lethagen et al. 2000). Whereas some studies have indicated that DDAVP is not effective in patients with thrombocytopenia or Glanzman disease, other patients with Glanzman disease or thrombocytopenia have been reported to respond (Mannucci et al. 1986; DiMichele and Hathaway 1990).
Toxicity
Most side-effects of DDAVP are minor. Facial flushing, often marked, and minimal elevation in pulse rate or blood pressure are observed, more frequently with the intravenous than with the subcutaneous or intranasal routes. The most common, clinically significant adverse event is hyponatraemia, which results from the antidiuretic effect of this vasopressin analogue. Hyponatraemic seizures have been reported in children aged 1 month to 8 years, especially when hypotonic intravenous fluids and multiple doses of DDAVP are administered concurrently in the surgical setting (Sutor 2000). Less severe but significant symptoms of headache, nausea or lethargy have been reported in adults after intranasal or repeated intravenous and subcutaneous administration (Dunn et al. 2000). Careful monitoring of i.v. fluids, urine output and electrolytes is therefore important, especially for children who receive DDAVP perioperatively, and in older patients when mild renal insufficiency decreases their ability to excrete free water. Patients should be instructed to restrict fluid intake for 24 h.
Isolated cases of thrombosis such as myocardial infarction, cerebral thrombosis and unstable angina have been reported after use of DDAVP in patients at risk for thrombotic events. In the surgical setting, one randomized, placebo-controlled study specifically designed to detect deep venous thrombosis in 50 patients undergoing hip surgery did not detect an increased incidence of thrombosis following DDAVP therapy (Flordal et al. 1992). However, given the suggestion that this prothrombotic agent increases the risk of myocardial infarction in cardiac surgery, it seems prudent to evaluate patients receiving DDAVP for possible occult coronary artery disease and to avoid the concurrent use of antifibrinolytic agents in these patients under most circumstances.
Lysine Analogue Antifibrinolytic Agents
Fibrinolysis occurs when plasmin generated from the proenzyme plasminogen by plasminogen activators digests fibrin clots (Collen 1999). Both plasmin and plasminogen bind to fibrin through specific lysine binding sites. The synthetic lysine analogues, tranexamic acid (AMCA) and epsilon-aminocaproic acid (EACA), delay fibrinolysis by reducing the binding of plasminogen to fibrin that is required for activation by plasminogen activators (Mannucci 1998). These agents have been available for more than 30 years to inhibit fibrinolysis and ensure clot stability (Dunn and Goa 1999).
AMCA was developed within a few years of EACA. AMCA is 10-fold more potent on a molar basis, and tends to cause less gastrointestinal discomfort at equivalent antifibrinolytic doses in healthy volunteers (Verstraete 1985). Much of the early use of antifibrinolytics in the US involved EACA, whereas AMCA was used relatively more frequently in Europe. Although AMCA and EACA have been considered equivalent agents, optimal dosing regimens have not been determined for either agent and reported toxicities vary (Porte and Leebeek 2002; McCormack 2012).
Lysine analogue antifibrinolytic drugs are effective and clearly indicated in rare inherited conditions associated with excessive fibrinolysis such as congenital α2-antiplasmin deficiency and hereditary angioedema (Aoki et al. 1979). However, much of the enthusiasm for these drugs has focused on acquired disorders with evidence of excessive systemic fibrinolysis, especially cardiac bypass surgery and, to a lesser degree, orthopaedic surgery performed with the use of tourniquets. Both agents are useful when administered topically in areas with excessive local fibrinolysis such as the oral cavity and uterine cavity. In these instances, efficacy has been well established, and side-effects are minimal compared with systemic administration (Nilsson 1975, 1980; Mannucci 1998; Konig et al. 2013). AMCA and EACA are also administered to reduce bleeding in such conditions as amegakaryocytic and peripheral immune-mediated thrombocytopenia, where the indication is less obvious, and efficacy is presumably related to stabilization of fibrin clots (Gardner and Helmer 1980; Bartholomew et al. 1989; Fricke et al. 1991).
Dose and Administration
Both agents may be administered orally, intravenously or topically. EACA and AMCA are well absorbed orally, and are cleared virtually unchanged by the kidney (Verstraete 1985). The drugs are distributed widely throughout the body. AMCA crosses into cerebrospinal fluid, semen, synovial fluid and cord blood, but is not secreted in saliva (Dunn and Goa 1999). In the non-operative setting, the half-life for both agents is approximately 1–2 h in patients with normal renal function. The dose should be reduced in renal failure (Dunn and Goa 1999). A typical total daily oral dose for EACA is 10–24 g, administered as 2–4 g every 3–4 h, and for AMCA 3–4 g, administered as 1 g every 6–8 h. Dosage in individual studies varies widely. Studies have attempted to optimize dosing for both agents in conditions such as cardiac surgery where the clearance and distribution of these agents is altered (Butterworth et al. 1999, 2001; Dowd et al. 2002).
Indications
A randomized, double-blind placebo-controlled study of AMCA administered to reduce bleeding in 38 patients with acute myeloid leukaemia found a significant reduction in bleeding episodes and platelet transfusions during consolidation, but not during induction therapy (Shpilberg et al. 1995). A smaller study of eight patients with amegakaryocytic thrombocytopenia (seven with severe aplastic anaemia and one with myelodysplasia) treated with AMCA in a placebo-controlled, double-blind, crossover design found no such reduction in this setting. Bleeding increased in patients while receiving AMCA (Fricke et al. 1991).
Lysine analogues are often used in cardiac bypass surgery, where excessive bleeding and fibrinolysis are associated with extracorporeal devices. Although early studies did not reveal a uniformly beneficial effect, antifibrinolytic use has increased as repeated analyses found that EACA prophylaxis significantly reduces postoperative blood loss and re-exploration, without increasing thromboembolic events (Munoz et al. 1999). In contrast with DDAVP, lysine analogues are administered prior to bypass. A meta-analysis of placebo-controlled studies of antifibrinolytic agents in cardiac surgery found that all antifibrinolytic agents were effective in reducing blood loss and transfusions, but no agent reduced mortality; neither did they increase the risk of stroke, myocardial infarction, or renal failure (Brown et al. 2007). Antifibrinolytic agents have been shown to reduce bleeding in total knee arthroplasty where use of a tourniquet produces excessive fibrinolysis (Hiippala et al. 1997), and to correct laboratory evidence of fibrinolysis in liver transplantation (Kaspar et al. 1997; Dalmau et al. 2004). Transfusion requirements were not reduced in these settings.
Other uses of lysine analogues include those based on well-designed studies as well as anecdote, with the strength of evidence often unrelated to clinical enthusiasm. Two retrospective uncontrolled studies of 31 patients with thrombocytopenia, including amegakaryocytic and immune-mediated aetiologies, demonstrated benefit with oral EACA (Garewal and Durie 1985; Bartholomew et al. 1989). Although not widely used for control of gastrointestinal haemorrhage, in which fibrinolytic enzymes may in fact play a role, AMCA has been associated with a reduced incidence of rebleeding and a 30–40% reduction in mortality in a meta-analysis that included 1200 patients from double-blind placebo-controlled trials (Henry and O’Connell 1989). A similar analysis found significant improvement in women with menorrhagia who received antifibrinolytic agents compared to hormonal methods and other modalities (Lethaby et al. 2000). An oral tablet has been formulated for this indication. AMCA reduced the incidence of rebleeding in patients with subarachnoid haemorrhage in three double-blind placebo-controlled studies subjected to Cochrane analysis (Roos et al. 2003). However, based on 1041 patients in three trials, antifibrinolytic treatment did not evidence improved outcome. Treatment increased the risk of cerebral ischaemia in five trials (Roos et al. 2003). Uncontrolled observations have proposed that shorter courses of high-dose EACA, or lower dose AMCA, might reduce rebleeding and avoid the complications associated with long-term use in patients scheduled for early aneurysmal therapy to control subarachnoid hemorrhage. Topical or local administration of the lysine analogues has been successful in a variety of clinical settings. Topical AMCA dramatically reduced the incidence of rebleeding after traumatic hyphaema, decreased systemic symptoms compared to oral administration and improved long-term outcomes compared with untreated control subjects (Crouch et al. 1997). Similarly, topical tranexamic acid (1 g in 100 ml of saline) administered during open heart surgery directly into the pericardial cavity and over the mediastinal tissues before closure reduced chest tube drainage, but not transfusion requirements, without detectable AMCA blood levels (De Bonis et al. 2000).
Antifibrinolytic therapy would seem well-suited to reduce massive haemorrhage, whether from high-risk surgical procedures or from trauma. A large three-arm randomized trial (BART trial) comparing the effects of different agents on blood loss and mortality in high-risk cardiac surgery patients had to be terminated early when one of the agents, aprotinin, was associated with an unacceptably high mortality when compared with the lysine analogues (see below) (Fergusson et al. 2008). A review of the trauma literature in 2004 concluded that there is insufficient evidence from randomized controlled trials of antifibrinolytic agents in trauma to either support or refute the use of these drugs (Coats et al. 2004). Since then, a large, global, multicenter, randomized, double blind, placebo controlled trial (CRASH-2) studied the effects of early administration of a short course of AMCA on death, vascular occlusive events, and the receipt of blood transfusion in some 20 000 adult trauma patients at risk for significant haemorrhage. (CRASH-2 collaborators 2010) CRASH-2 concluded that overall mortality and mortality related to bleeding both were reduced in the AMCA cohort. The conclusions of this potentially important study are muddied by several defects in study design, execution and detail. There were 274 participating hospitals, most in the developing world, but no information regarding specific site enrolment, the standard of patient care, injury assessment scoring, or blood availability. Many enrolled patients did not meet enrolment criteria. In terms of safety, it is surprising that no severe adverse events were reported, nor were clinical or laboratory criteria for detection of thrombotic events detailed. There was an unexpected and unexplained reduction in risk of myocardial infarction in patients receiving AMCA compared with placebo. CRASH-2 would not likely be sufficient basis for approval of AMCA for use in trauma.
The topical use of antifibrinolytic agents to control bleeding in dental surgery for patients with haemophilia and other conditions predisposing to excessive haemorrhage including oral anticoagulation deserves special mention. In this setting, antifibrinolytic treatment probably counteracts excessive fibrinolysis in the oral cavity, which lacks endogenous fibrinolytic inhibitors (Sindet-Pedersen et al. 1989). More than 30 years ago, controlled trials demonstrated reduced bleeding and decreased requirements for clotting factor concentrates in patients with haemophilia A and B who received systemic oral administration of EACA, 6 g four times daily for 7–10 days (Walsh et al. 1971, 1975). More recently, AMCA mouthwash (10 ml of a 4.8% aqueous solution applied prior to sutures and as a 2-min rinse four times a day for 7 days) dramatically reduced bleeding episodes after dental procedures (Sindet-Pedersen et al. 1989). Topical administration of AMCA, along with the use of DDAVP and fibrin sealant, has become standard treatment in haemophilia dental centres.
Toxicity
The most common side-effects of lysine analogue antifibrinolytic therapy are mild dose-dependent gastrointestinal symptoms, such as nausea, cramping and diarrhoea. These symptoms are less frequently reported with AMCA than with EACA (Verstraete 1985; Dunn and Goa 1999). Rhabdomyolysis, possibly due to inhibition of carnitine synthesis, has been described, although rarely, when oral EACA has been continued beyond 4 weeks (Seymour and Rubinger 1997). Rhabdomyolysis has not been reported after AMCA administration. Some patients who have developed this complication while on EACA have been successfully treated for many months with AMCA (Dunn and Goa 1999).
When administered at usual clinical doses, the antifibrinolytic drugs do not appear to differ in their potential to cause thrombosis. Widespread thrombosis has occurred in the setting of disseminated intravascular coagulation (DIC), where increased fibrinolysis may protect against organ ischaemia. Some studies have attempted to pair antifibrinolytic therapy with heparin anticoagulation in DIC and ‘excessive’ fibrinolysis characterized by low levels of α2-antiplasmin. Although bleeding decreased in most cases, some patients still developed generalized organ system failure attributed to thrombosis (Williams 1989). High-grade ureteral obstruction due to inhibition of urokinase and clot formation may occur when antifibrinolytics are used to treat patients with haematuria (Schultz and van der Lelie 1995). Topical therapy, administered by irrigation, has been successful for control of intractable urinary bleeding localized to the bladder (Singh and Laungani 1992).
Aprotinin (Trasylol)
Aprotinin is a bovine lung-derived 58-residue polypeptide with broad antiprotease activity directed towards trypsin, kallikrein, thrombin and plasmin (Longstaff 1994). Aprotinin activity is ordinarily expressed in kallikrein inactivator units (KIUs). However, the relationship of this activity to the mechanism(s) of its haemostatic action is not clear. Aprotinin has potent antifibrinolytic activity, at least as regards circulating plasmin, and this is believed to be the primary haemostatic mechanism (Ray and Marsh 1997). Other proposed mechanisms for the haemostatic function involve a long-disputed platelet-protective effect and inhibition of protease-activated receptors found on platelets, leucocytes and vascular endothelium (Shigeta et al. 1997; Mannucci 1998). Aprotinin does not induce platelet adhesion or aggregation, but inhibits various platelet agonists produced during surgical procedures and cardiopulmonary bypass. Aprotinin also inhibits complement activation, attenuates the release of tissue necrosis factor-alpha (TNF-α), IL-6 and IL-8 and inhibits endogenous cytokine-induced nitric oxide synthase induction (Mahdy and Webster 2004).
Dosage and Clearance
As was the case with the other antifibrinolytic agents, aprotinin has been used in different doses and regimens; the most common regimen involved a loading dose of 2 million KIU followed by continuous infusion of 500 000 KIU/h (Royston et al. 1987). After i.v. injection, aprotinin distributes into the extracellular space, leading to a rapid decrease in plasma concentration. The drug is eliminated in a biphasic pattern: a rapid-phase half-life of approximately 40 min and a slower phase half-life of 7 h (Porte and Leebeek 2002).
Indications
During trials of aprotinin designed to reduce neutrophil activation in patients undergoing repeat open heart surgery, investigators noted that the operative field was dry and that transfusion requirements decreased eight-fold compared with control patients who received no drug (Royston et al. 1987). Subsequent trials demonstrated the effectiveness of aprotinin in patients undergoing procedures requiring cardiopulmonary bypass (Lemmer et al. 1994), particularly in operations characterized by large volume blood loss (Prendergast et al. 1996; Smith 1998, Fergusson et al. 2008). Meta-analyses have confirmed these findings (Levi et al. 1999; Munoz et al. 1999; Henry et al. 2011).
Adverse Events
Anaphylaxis, not unexpected with a bovine-derived protein, has been reported in approximately 0.5% of initial exposures and up to 9% of repeat exposures (Diefenbach et al. 1995; Scheule et al. 1997). Mild hypersensitivity reactions are more common. Quantitative detection of anti-aprotinin IgE and IgG have limited predictive value for identifying patients at risk for anaphylaxis following re-exposure to aprotinin, and repeat treatment within 6 months is discouraged (Dietrich et al. 1997, 2001). However neither the clinical trial database that led to approval of the drug in the US nor the numerous randomized, controlled trials conducted after approval identified any association between aprotinin and a short-term increase in the risk of death, cardiovascular events, or any serious renal toxic effects (Lavee et al. 1993; Bidstrup et al. 1993; Havel et al. 1994; Lemmer et al. 1994). Further, imaging studies did not identify an increased risk of deep vein thrombosis in the setting of orthopaedic surgery (Murkin et al. 2000; Samama et al. 2002). Still concerns regarding the possibility of unintended thrombosis, particularly myocardial infarction, bypass graft occlusion and deep vein thrombosis have persisted for more than 20 years (Bohrer et al. 1990; Cosgrove et al. 1992). Several more recent observational studies as well as additional data supplied by the drug manufacturer raised serious concerns regarding renal toxicity (Mangano et al. 2006; Hiatt 2006). A randomized trial comparing lysine analogs with aprotinin was terminated early when an interim analysis detected increased deaths from cardiac causes in the aprotinin arm (Fergusson et al. 2008). Concerns regarding cardiac, renal, and cerebrovascular toxicity have led the US FDA to remove aprotinin from the US market.
Reversal of Warfarin Anticoagulation
Vitamin K and Its Antagonists (Warfarin, Bishydroxycoumarin, Dicumoral®)
Henrik Dam (Dam and Glavind 1938) discovered that chicks fed an ether-extracted diet developed a haemorrhagic diathesis that responded to a fat-soluble factor that became known as Koagulationvitamin or vitamin K. Vitamin K was subsequently found to correct the acquired bleeding tendency during the first week of life that has come to be known as haemorrhagic disease of the newborn (Brinkhous and Smith 1937; Dam and Dyggve 1952). With recognition that the bleeding in ‘sweet clover disease of cattle’ was caused by a compound identified later as bishydroxycoumarin, or Dicumoral®, which interfered with vitamin K-dependent synthesis of active prothrombin, vitamin K acquired a prominent role in the management of excessive anticoagulation due to warfarin, superwarfarin rodenticides and other orally active anticoagulants with similar mechanisms of action. Vitamin K is also indicated in acquired vitamin K deficiency that results from the combination of poor diet and antibiotic therapy that occurs commonly in intensive care settings (Alperin 1987; Cohen et al. 1988).
Vitamin K1 is synthesized by plants, possessing the same phytyl side-chain as chlorophyll, and is the major dietary source of vitamin K. The vitamin K1 content varies widely between foods, yet diet is a trivial source of variation in patient response to oral anticoagulants (Schurgers et al. 2004). Absorption of dietary vitamin K occurs in the ileum and requires the formation of mixed micelles composed of bile salts and the products of pancreatic lipolysis. Oral absorption is impaired in conditions of either biliary obstruction or pancreatic insufficiency. Vitamin K2 (menadione) is synthesized by bacterial flora of the large intestine and appears to have a reserve function to protect against dietary vitamin K deficiency, a rare occurrence in healthy adults (Vermeer and Schurgers 2000).
Mechanism of Action
Vitamin K functions as an essential cofactor for the post-translational gamma carboxylation of glutamic acid moieties in the N-terminal region of a series of proteins (Furie et al. 1999). Synthesis of functional prothrombin, as well as of coagulation factors VII, IX, X and naturally occurring anticoagulant proteins C, S and Z, requires vitamin K (Vermeer and Schurgers 2000). The decarboxylated, functionally inactive forms of vitamin K-dependent proteins (also called proteins induced by vitamin K absence or PIVKA) may be detected in the circulation of patients on oral anticoagulants or in those with vitamin K deficiency due to malabsorption. The enzyme(s) that reduce and recycle vitamin K and vitamin K epoxide have different sensitivities to oral anticoagulant-induced inhibition, which explains why vitamin K functions as an antidote to excessive oral anticoagulation and why patients on warfarin therapy who receive high doses of vitamin K may appear resistant to re-institution of therapy (Furie et al. 1999). The primary site of inhibition by warfarin is vitamin K epoxide reductase, which under physiological conditions is also the enzyme that reduces vitamin K to the active hydroquinone required for carboxylation. However, a second, warfarin-insensitive reductase can reduce vitamin K to the active hydroquinone in the presence of high tissue concentrations of vitamin K. Thus, exogenous vitamin K can produce additional active vitamin K hydroquinone via this warfarin-insensitive step, bypassing the warfarin-induced inhibition of vitamin K epoxide reductase and reversing excessive anticoagulation (Furie et al. 1999). If vitamin K levels accumulate, this same process may also lead to warfarin resistance upon resumption of anticoagulation.
Lowered levels of vitamin K-dependent clotting factors of patients taking oral anticoagulants recover at the same rate during the initial 8 h following vitamin K administration, but subsequent recovery depends on the rate of synthesis of new gamma carboxylated factors by the liver. Levels of factor VII reconstitute most rapidly, followed by factors IX and X, and lastly by prothrombin (van der Meer et al. 1968). The differential rate of circulating factor levels after vitamin K administration has practical importance. Oral anticoagulation is most frequently monitored using the prothrombin time, which is most sensitive to decreases in factor VII levels. During stable anticoagulant therapy, the several vitamin K-dependent coagulant and anticoagulant proteins are decreased in relatively constant ratios to one another. However, administration of vitamin K for excessive anticoagulation may result in a prothrombin time correction related largely to a rapid increase in the factor VII level and underestimating the bleeding risk from the lower levels of factors IX, II and X. Test correction and clinical bleeding do not necessarily correlate closely (Crowther et al. 2009, and see below).
Dose and Administration
Both vitamins K1 and K2 are active when administered orally or intravenously. Liquid preparations are prepared as a colloid solution, each millilitre containing 2 or 10 mg of phytonadione in a polyoxyethylated fatty acid derivative, which functions as a cremaphor to solubilize the vitamin K. Intramuscular administration may cause haemorrhage in anticoagulated patients and should be reserved for prophylaxis against haemorrhagic disease of the newborn (1 µg). The side-effects reported with intravenous administration (see below) have limited its use, even though intravenous administration is more rapid and reliable than the subcutaneous route (van der Meer et al. 1968; Nee et al. 1999). Scored tablets are available, containing 5 mg of phytonadione, permitting a dose range as low as 2.5 mg. Special formulations have been prepared for studies with lower doses. Although injectable and oral preparations have been available for nearly half a century, the appropriate dose and route of administration remain controversial (Taylor et al. 1999).
The response to vitamin K administration depends on the direction, rate of change and degree of prolongation of clotting times, the dose and type of anticoagulant administered and the presence of concurrent liver disease, antibiotics or dietary factors (Cosgriff 1956; Schulman 2003) Anticoagulant reversal is more prompt and more reliable with intravenous than with subcutaneous administration of vitamin K (Nee et al. 1999; Raj et al. 1999). In a prospective, randomized, single-blind study of 22 patients, the INR decreased from a mean initial value of 8.0 to 4.6 and 3.1 after 8 and 24 h respectively in patients who received 1 mg intravenously, compared with a mean initial value of 8.5, which decreased to 8.0 and 5.0 at the same intervals in patients who received 1 mg subcutaneously (Raj et al. 1999). The response after oral ingestion of vitamin K is more rapid than that observed after simple discontinuation of warfarin, which can be prolonged in the elderly, in malnourished patients, and in those with liver disease (Garcia et al. 2006). A single low-dose oral vitamin K tablet (1–2.5 mg) effectively reduces the INR in otherwise stable over-anticoagulated patients within 24 hours of administration (Ageno et al. 2005). Oral vitamin K doses as low as 0.5–2.5 mg (Crowther et al. 2000; Gunther et al. 2004) have been recommended for patients at risk with mild to moderate prolongation of the INR, but who are not bleeding.
Vitamin K deficiency occurs commonly in malnourished patients in intensive care units, either with or without concomitant antibiotic use, accounting for four or five cases yearly in one tertiary care referral centre (Alperin 1987; Cohen et al. 1988). Administration over 30 min of 5–25 mg of intravenous vitamin K corrected the coagulopathy within 12 h (Alperin 1987). A much smaller dose of vitamin K, 1 mg intravenously, is effective within 24 h when vitamin K deficiency is due to malabsorption in patients who are not critically ill, and the effect lasts 1–2 weeks (van der Meer et al. 1968).
Clinical Reversal of Excessive Anticoagulation
Oral anticoagulation with vitamin K antagonists such as warfarin is ordinarily monitored with the prothrombin time using the International Normalized Ratio (INR). When excessive oral anticoagulation occurs in the absence of bleeding (INR < 5.0), omission of a dose or two of anticoagulant may suffice until the therapeutic range is reached. With a more prolonged INR (>5.0, but <9.0), correction may be achieved more rapidly if a small dose of vitamin K is administered, most conveniently as a 1-mg oral dose (Garcia et al. 2006). This approach may also reduce the risk of minor haemorrhage for patients with INR values of between 4.5 and 10 (Crowther et al. 2000; Wilson et al. 2004). Alternatively, 0.5 mg of vitamin K1 may be injected intravenously. Higher doses should be avoided as they may lead to overcorrection and resistance to anticoagulation with vitamin K antagonists for several days. Subcutaneous injection is not recommended; absorption by this route is variable. If more rapid reversal is required, for example to permit urgent surgery, vitamin K (≤5 mg) can be administered orally with the expectation that a reduction of the INR will occur in 24 h. Despite more rapid correction of INR, low-dose therapy may not prevent bleeding in stable adults with excessive anticoagulation and no other bleeding risks. In a randomized placebo-controlled trial to correct excessive anticoagulation in 724 outpatients in 14 centers in Canada, the U.S. and Italy, oral vitamin K (1.25 mg) did not reduce bleeding in warfarin recipients with INRs between 4.5 and 10.0 (Crowther et al. 2009). The study highlights the distinction between correcting a laboratory value and preventing haemorrhage. However this trial is underpowered to detect major bleeding episodes, and does not indicate whether slightly larger oral doses of vitamin K (e.g. 2.5–5.0 mg) might do so. It still seems prudent to administer vitamin K to excessively anticoagulated patients with who have additional reasons to be at increased risk of bleeding.
For excessive anticoagulation associated with bleeding, especially with an INR dangerously outside of the therapeutic range, and particularly in the presence of intracranial haemorrhage, administration of vitamin K1 is not sufficient; full reversal will not occur for 12 to 24 h. In patients with serious bleeding and elevated INR, regardless of the magnitude of the elevation, prudent management includes discontinuing warfarin therapy and administering vitamin K (10 mg) by slow IV infusion supplemented with fresh frozen plasma, prothrombin complex concentrate (PCC), or recombinant factor VIIa, depending on the urgency of the situation (Ansell et al. 2008). Vitamin K should be repeated every 12 h for persistent INR elevation.
For the less urgent situations, FFP is ordinarily the drug of choice, but because of isohemagglutinins, blood group specific plasma should be used. The volume of plasma required for reversal may be large, and if volume overload is a problem, prothrombin complex concentrate is effective (Makris et al. 1997). Prothrombin complex has the additional advantages of specific factor replacement and treatment to inactivate pathogens. Yasaka et al. have reported that a dose of 500 IU of PCC was optimal for rapid reversal for an INR of <5.0 and that higher doses are probably needed for more elevated INRs (Yasaka et al. 2005).
However preparations of PCC differ in the composition of clotting factors depending upon the manufacturer and some are relatively deficient in F VII. When plasma or prothrombin complex is used, the volume (or units) may be calculated as below (Schulman 2003):
Related posts:
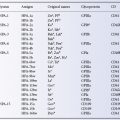 Immunology of leucocytes, platelets and plasma components
Immunology of leucocytes, platelets and plasma components
 Red cell antibodies against self-antigens, bound antigens and induced antigens
Red cell antibodies against self-antigens, bound antigens and induced antigens
 ABO, H, LE, P1PK, GLOB, I and FORS blood group systems
ABO, H, LE, P1PK, GLOB, I and FORS blood group systems
 The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
 The Rh blood group system (including LW and RHAG)
The Rh blood group system (including LW and RHAG)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


